

Abstract
The human polyomavirus BK virus (BKV) is an important opportunistic pathogen whose disease prevalence continues to increase with the growing immunocompromised population. To date, the major determinant of replication in cell culture has not been formally proven. BKV exists as archetype virus and rearranged variants, which are classified based on the DNA sequence of their non-coding control regions (NCCRs). The archetype BKV NCCR is divided into five blocks of sequence and rearranged variants contain deletions and duplications of these blocks. In this study, a genetic system was developed and used to identify the major determinant of replication ability in primary renal proximal tubule epithelial cells, the natural host cell of BKV. This system was also used to analyze NCCR variants isolated from an immunocompromised patient which contain assorted rearrangement patterns and functional differences. This study solidifies the NCCR as the major genetic determinant of BKV replication ability in vitro.
Keywords: BKV, NCCR, HIV/AIDS
Introduction
BK virus (BKV) is a highly prevalent human polyomavirus originally isolated from the urine of a renal transplant patient with ureteric obstruction (Gardner et al., 1971). BKV is believed to be spread asymptomatically by a respiratory route, based on evidence that seroconversion takes place in early childhood and that BKV has been detected in tonsillar and salivary gland tissues (Goudsmit et al., 1982; Jeffers et al., 2009). However, data also suggest that a fecal-oral route of transmission may be possible (Vanchiere et al., 2009; Vanchiere et al., 2005a; Wong et al., 2009). The seroprevalence of BKV in the population is reached by the age of ten and is estimated to be between 65–90% (Kean et al., 2009; Knowles, 2006). Following primary infection, BKV establishes a lifelong, persistent, subclinical infection in the urinary tract (Chesters et al., 1983; Heritage et al., 1981). Renal tubule epithelial cells are a major site in which BKV persists and can be reactivated (Knowles, 2001; Moens et al., 2001). BKV is not known to cause disease in healthy, immunocompetent people, but it can be periodically isolated from their urine as a result of transient reactivation events (Knowles, 2001). In some clinical settings, BKV can reactivate and cause diseases such as hemorrhagic cystitis (HC) in bone marrow transplant patients and polyomavirus-associated nephropathy (PVAN) in kidney transplant patients. BKV can also reactivate in other immunosuppressed populations such as pregnant women and patients with HIV/AIDS (Doerries, 2006). Studies have shown both increased incidence and amount of virus present in the urine of HIV-infected patients as compared to uninfected people (Knowles et al., 1999; Markowitz et al., 1993). Although rare, BKV infection in HIV/AIDS patients can result in death (Cubukcu-Dimopulo et al., 2000).
BKV has a small double-stranded circular DNA genome that is divided into three genetic regions: the early coding region, the late coding region, and the non-coding control region (NCCR). BKV is classified into archetype virus and rearranged variants, based on the structure of the NCCR, which contains the origin of DNA replication and promoters for both the early and late coding regions. Archetype BKV is characterized by five blocks of DNA sequence within the NCCR; O, P, Q, R, and S (Fig. 1A). The O block contains the origin of replication and putative transcription factor binding sites. Rearranged variants of BKV are characterized by deletions and duplications in the sequence blocks that enhance replication ability of the virus and allow it to grow readily in cell culture. The early and late gene promoters and transcriptional enhancers, as defined in rearranged variants, are located in the P, Q, R, and S blocks (Markowitz et al., 1988; Moens et al., 1995; Moens et al., 2005). BKV archetype virus can be isolated from both healthy individuals and diseased patients, and it currently cannot be grown in cell culture. Rearranged BKV variants are most commonly isolated from patients with BKV diseases such as PVAN and HC, but not in all such patients, and replicate robustly in culture.
Fig. 1.

The NCCR determines replication efficiency of BKV. (A) Schematic of the swap genome with the archetype virus (Dik) NCCR. SpeI and SacII sites were inserted into the pBR322-Dunlop or –Dik vectors flanking the majority of the NCCR and a PmlI site was inserted between the early and late regions. (B) RPTE cells were transfected with recircularized viral genome and low molecular weight DNA was harvested 5 dpt. Samples were linearized, digested with DpnI, and analyzed by Southern blotting. The left panel shows Dik and Dunlop wt replication compared to the swap vectors containing the three inserted restriction enzyme sites, Dik3 and Dun3 respectively. The right panel shows all possible swap combinations. Each construct is designated by a three letter abbreviation. A=archetype and R=rearranged. The first letter denotes the NCCR; second letter, early region; third letter, late region. Marker, HindIII digest of pGEM-TU; Mock, mock transfection.
Previous studies have shown that when archetype virus, isolated directly from patient urine, is passaged for prolonged periods in cell culture, it accumulates rearrangements in the NCCR (Rubinstein et al., 1991). Likewise, although the archetype virus is the prevalent form of BKV, when high titers of virus are present in the serum of a patient with BKV disease, the appearance of rearranged variants increases and these eventually replace archetype as the predominant form of the virus (Gosert et al., 2008). This is likely due to increased replication efficiency of rearranged variants, resulting in the ability to outcompete archetype virus in an immunocompromised host. The dominance of rearranged variants in a host may directly relate to clinical outcome, since these variants are able to replicate more efficiently and show increased cytopathology in vitro (Gosert et al., 2008). A recent study showed that multiple rearranged quasispecies can be found in the urine or allograft biopsies of kidney transplant recipients, even within a single patient (Olsen et al., 2009). In addition to differences in the NCCRs between archetype virus and rearranged variants, it is possible that polymorphisms in the early and late coding regions can also contribute to replication ability. Comparing the Dik archetype virus (Goudsmit et al., 1981) and the Dunlop rearranged variant (Seif et al., 1979), for example, there are four amino acid differences in the early coding region: three in TAg and one in tAg. There also are nine amino acid differences in the late coding region: three in VP1 and six in VP2, three of which overlap in VP3. However, contributions of the NCCR, the early and late coding regions to replication have not been systematically examined.
For the present study, a swap vector system was developed to resolve the major determinants of BKV replication in primary renal proximal tubule epithelial (RPTE) cells. This system allows for independent analysis of the major viral DNA functional regions, the NCCR and the early and late coding regions, from different BKV isolates. Using this system, we were able to formally prove that the NCCR is the major determinant of replication in cell culture. The swap vector system was then used to functionally analyze variants isolated from the urine of an AIDS patient with BKV viruria. These clones displayed a series of NCCR rearrangements with variable levels of replication ability and infectious progeny production. These studies provide a useful genetic system with which to analyze patient isolates and dissect specific determinants of BKV replication.
Results
The NCCR is sufficient to determine replication ability in cell culture
BKV variants with rearranged NCCRs are able to replicate in cell culture while viruses with an archetype NCCR are unable to do so. To determine whether other regions of the BKV genome contribute to replication ability, we created a swap vector system that allows for the exchange of the early coding region, late coding region and the NCCR between archetype virus and rearranged variants (Fig. 1A). Unique restriction sites were introduced into the archetype (Dik) and rearranged (Dunlop) variant genomes to create the swap vectors and investigate the contributions of the three regions to the replication ability in RPTE cells. SpeI and SacII sites were introduced into regions with no sequence variation between Dik and Dunlop in the O block and the S block, respectively, flanking the majority of the NCCR. Additionally, a PmlI site was inserted between the early and late regions in both the archetype and rearranged genome in a pBR322 backbone (Seif et al., 1979). The swap vectors were named Dik-3-site (Dik3), and Dunlop-3-site (Dun3), in reference to the insertion of the three restriction sites. Using these vectors, we constructed all possible combinations of the three regions of the archetype virus and rearranged variants. We were then able to assay replication ability by a DpnI resistance assay, which distinguishes methylated transfected plasmid DNA that has not been replicated in eukaryotic cells from unmethylated DNA that has been replicated in eukaryotic cells (Pipas et al., 1983).
Each recombinant genome was excised from the plasmid, re-circularized, and transfected into RPTE cells. At five days post-transfection, low molecular weight DNA was isolated (Hirt, 1967), linearized, digested with DpnI, and assayed by Southern blotting (Fig. 1B). The exogenous restriction sites had no impact on the replication ability of the genome (compare lanes 3 to 4 and lanes 5 to 6). Only those recombinants created containing the rearranged variant NCCR were able to replicate efficiently (lanes 8, 9, and 11) as compared to recombinants containing the archetype NCCR (lanes 7, 10, and 12). Of note, a low level of archetype replication was seen in lanes 3, 4, 7, 10, and 12; however no infectious progeny were detected by an infectious unit assay (Figure 3C and data not shown). What appears to be low level archetype virus replication could have been the result of incomplete DpnI digestion of input DNA. These data demonstrate that the NCCR is the major determinant of the ability to replicate in RPTE cells.
Fig. 3.

Clones isolated from a clinical specimen display variable replication abilities. RPTE cells were transfected with recircularized viral genomes. (A, B) Low molecular weight DNA was harvested 4 dpt and analyzed as in Fig. 1. (A). Representative Southern blot. The two panels are different exposures of the same blot. Arrow indicates the DpnI-digested band used for normalization among samples. Mock, mock transfection. (B) Quantification of replication data from three independent experiments, error bars represent standard error of the mean.. (C) Viral lysates were prepared from parallel transfections at 7 dpt and assayed for infectious progeny as described in the Materials and Methods. Results are from three independent experiments, error bars represent standard error of the mean. The * represents a measurement below the limit of detection.
Characterization of BKV NCCR variants isolated from an immunocompromised patient
To test the utility of this system for analyzing patient isolates, we cloned the BKV NCCR from a urine sample of an AIDS patient with BKV viruria. The serum and the urine of this patient were tested for the presence of BKV and JCV by quantitative PCR as previously described (McNees et al., 2005). JCV was not detected in either fluid and BKV was only detected in the urine, at 150 genomes/mL. The BKV amplicons from the patient were sequenced and classified as genotype Ia based on VP1 polymorphisms (Jin et al., 1993; Zheng et al., 2007). We were able to isolate five distinct NCCRs from this single urine specimen displaying differing degrees of rearrangement (Fig. 2A). These clones were designated TCH for their location of isolation (Texas Children’s Hospital) and numbered 1–5. Clones TCH3 and TCH4 have a partial P block inserted before a complete P block, and differ from each other by the presence of a point mutation. Clones TCH1 and TCH2 each have unique truncations in their R blocks. In addition, TCH1 is completely missing the S block. The most highly rearranged clone, TCH5, contains a partial duplication of both the P and the Q blocks inserted between the full P and Q blocks. All five also have a single nucleotide polymorphism in the O block as compared to both Dik and Dunlop.
Fig. 2.

(A) Schematic of the NCCRs of viruses isolated from a single clinical specimen. The NCCR is divided into blocks of sequence O, P, Q, R, and S. The block structures of the archetype virus (Dik), the rearranged variant (Dun), and the variants isolated from the patient are shown. The * represents the O block polymorphism and the † represents other mutations relative to the archetype P block sequence. The P′ and P″ blocks of Dunlop are variations of the P block. (B) A schematic of the Dik C65T and TCH2 T65C mutants created by site directed mutagenesis.
We first assessed the replication ability of each isolate by introducing the restriction enzymes SpeI and SacII into the NCCR of each variant and then swapping them into the Dun3 backbone. All genomes were excised from the plasmid backbone, re-circularized, and transfected into RPTE cells. Four days post-transfection, low molecular weight DNA was isolated and assayed for replication ability by Southern blotting (Fig. 3A). Day 4 was chosen because parental rearranged variant viral replication can be easily detected at this time point (data not shown). To quantify replication ability, the replicated genome band was normalized to a DpnI-digested input band (Fig. 3A, arrow) to control for transfection efficiency and the archetype virus (Dik3) was set to one (Fig. 3B). The clones show various genome replication abilities ranging from low and intermediate (TCH1, TCH3, TCH4, TCH2) to high (TCH5), which replicated to levels similar to Dun3.
To further characterize these patient isolates, infectious progeny production was assessed using an infectious unit assay as previously described (Low et al., 2004). Each recombinant genome was transfected as described above and cell lysates were harvested seven days post-transfection. Progeny production followed the same trend as replication, with low and intermediate level replicating clones producing less progeny than the high level replicating clone (Fig. 3C). These data indicate that NCCR rearrangements are able to account for functional differences assayed in vitro.
Finally, we determined whether the O block polymorphism (C65T where 1 is the O block starting position) affected replication. Site-directed mutagenesis was used to recreate the polymorphism common to all the patient variants in an archetype virus background (Fig. 2B). Additionally this polymorphism was reverted to the Dik sequence in a TCH2 background. Replication ability was assessed by DpnI resistance assay as above. Results from this assay show that the point mutation does not affect the replication ability of the viruses (Fig. 4).
Fig 4.
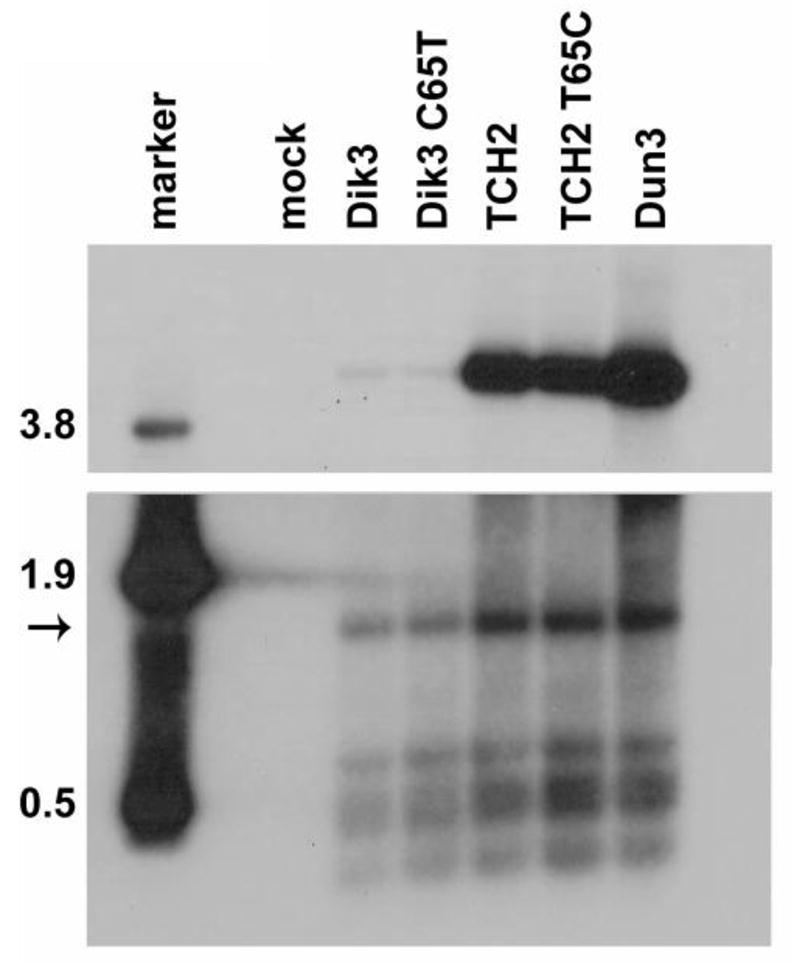
The O block polymorphism does not affect replication ability. The C65T mutation was created and reversed in the O blocks of Dik3 and TCH2, respectively, and the genomes were assayed as in Fig. 3A. The blot is representative of two independent repeats, and the two panels are different exposures of the same blot. Marker: HindIII digest of pGEM7-TU (size of bands in kb); Mock, mock transfection.
Discussion
The determinants that regulate the ability of archetype virus and rearranged variants of BKV to replicate in cell culture have not yet been systematically mapped. In human embryonic kidney and adult human skin fibroblast cells, which are not natural target cells for BKV, prolonged passage of archetype virus results in the accumulation of NCCR rearrangements leading to increased replication ability (Rubinstein et al., 1991). Thus, the NCCR was assumed to be the main determinant of viral replication based on the data that rearrangements are apparently selected for in cell culture; however, this research was performed in cells that are not the natural host of BKV infection. Furthermore the dependence of replication ability on the NCCR has not previously been directly proven. Additionally, there is suggestive evidence for the contribution of the early and late coding regions in regulating polyomavirus replication ability. For example, a naturally occurring Q169L mutation in the BKV large tumor antigen (TAg) protein has been proposed to stabilize its binding to DNA, which could conceivably affect replication ability since TAg stimulates viral replication (Smith et al., 1998). Of note, this mutation is not present in either the Dik or Dunlop viruses used in the present study.
In this study, we showed directly that the NCCR is the major determinant of BKV replication in cell culture. To answer the question of which region determines viral replication ability, we developed a swap vector system. This allowed us to create recombinants of the archetype virus and rearranged variant genomes to assess the roles of the NCCR, early coding region, and late coding region in replication. Insertion of restriction enzyme sites between the regions of the genome did not affect replication. We then used this vector system and demonstrated that the NCCR is the major determinant of viral replication ability in RPTE cells because only those recombinants containing a rearranged variant NCCR were able to replicate efficiently. This result seems logical considering that the NCCR, early and late coding regions all have functions that are required for viral replication, but the early and late coding regions are more genetically constrained. For example, genetic instability in TAg would be problematic because it needs to be able to bind the origin in the NCCR to initiate replication (Eichman et al., 2004). TAg also has many other functions that require interaction with host proteins, suggesting that the TAg sequence needs to be stable to maintain these interactions. The same restrictions may be seen for the late coding region since the capsid is a relatively rigid structure whose main component is the VP1 protein, which needs to be able to recognize host gangliosides for entry into the cell (Low et al., 2006).
We used the swap vector system to analyze BKV NCCRs cloned directly from a single urine specimen from an AIDS patient with BKV viruria. The NCCRs of five distinct isolates with various rearrangements were introduced into the Dunlop background in order to analyze replication ability and infectious progeny production. All five clones were able to replicate better than the archetype virus, Dik, and each contained at least partial duplications or deletions in the NCCR sequence blocks. In general, the isolates that had a greater degree of rearrangements had higher replication ability and infectious progeny production than those with fewer rearrangement. However, with such a small sample size it was difficult to discern a clear pattern of whole or partial block rearrangements that correlated to replication ability. Clones TCH3 and TCH4 differed by only a point mutation in the P block and displayed similar functional profiles. There was no archetype virus isolated from this patient’s urine but this does not confirm its absence: due to the limits of PCR-based assay detection, only the most highly represented variants are likely to be detected. The absence of archetype virus from this patient’s urine sample may indicate the severity of the patient’s immunocompromised state, however. The presence of rearranged BKV variants in serum is associated with increased replication and inflammation (Gosert et al., 2008). While virus excreted in the urine may be produced at a different site of replication from serum, it may still follow the same trend as the virus found in the serum with respect to clinical severity. Additionally, the HIV-1 Tat protein is able to activate the BKV early promoter in trans through sites located in the NCCR (Gorrill et al., 2006). This activation increases TAg expression and therefore, viral DNA replication. This is another possible factor influencing the reactivation of BKV in HIV-infected patients.
Further study is needed to determine why rearranged variants, and not archetype virus, are able to replicate in cell culture. Archetype BKV is more frequently isolated from patients than rearranged variants, but there are no reports of successfully culturing this form. It is possible that archetype virus and rearranged variants naturally replicate in separate populations of cells in the host, such that proximal tubule cells are permissive to rearranged variants but not archetype virus. Another possible, but not mutually exclusive, explanation is that the presence or absence of certain transcription factor binding sites in the NCCR of rearranged variants is required for replication in tissue culture. We were unable to identify any obvious pattern contributing to replication ability from an analysis of the variant NCCRs in comparison to the archetype virus using a transcription factor binding site prediction program, AliBaba2.1 (see http://www.gene-regulation.com/pub/programs.html).
In addition to NCCR rearrangements affecting viral gene transcription, it is possible that there are direct effects on DNA replication of the virus. It has been shown that there are three TAg binding sites within the SV40 regulatory region (i.e., the SV40 NCCR) that are assumed to be present in the BKV NCCR as well (Del Vecchio et al., 1989; Yang et al., 1979). Rearrangements in the SV40 regulatory region affect the binding stability of TAg to these sites, altering the DNA conformation and the levels of DNA replication (Wilderman et al., 1999). Also, cis-acting sequence elements in mouse polyomavirus and SV40 are involved in the activation of replication (de Villiers et al., 1984; O’Connor et al., 1988; Tang et al., 1987). Although the O block polymorphism C65T, which is located in putative TAg binging site I, is present in all of the BKV variants in the present study, this mutation did not significantly affect replication ability. We can use the swap vector system in the future to further characterize the role of the rearranged NCCR by dissecting its contribution to transcription and replication.
Materials and Methods
Patient and Sample Collection
Urine specimens were collected periodically from subjects enrolled in a prospective study of polyomavirus excretion in the urine of immune compromised patients. The study protocol was approved by the Institutional Review Board for Human Subjects Research at Baylor College of Medicine. Informed consent and assent, when appropriate, was obtained from subjects prior to specimen collection in 2001. The specimens in this report are from a single patient.
Cell Culture
Primary human RPTE cells were maintained for up to six passages in renal epithelial growth medium as previously described (Abend et al., 2007). Infectious unit assays were performed as previously described (Low et al., 2004).
Plasmids
The pBR322-Dunlop plasmid (ATCC #45025, pBKV) was received from Peter Howley (Seif et al., 1979). The pBR322-Dik plasmid was received from John Lednicky. Both of these genomes are inserted into the pBR322 backbone at the BamHI site. Site-directed mutagenesis was performed using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) as described previously (Abend et al., 2008) on both pBR322-Dik and Dunlop to create Dik-3-site (Dik3) and Dunlop-3-site (Dun3) swap vectors. The SpeI site was inserted into the O block using primer A and its reverse complement and the SacII site was inserted using primer B and its reverse complement (Table 1). The PmlI site was inserted between the early and late regions using primer C and its reverse complement. Swap combinations were created by digesting 10 μg of Dik3 and Dun3 with the appropriate restriction enzymes to obtain the desired fragment (NCCR, early, or late). Each fragment was then gel extracted and ligated with its counterpart genome using T4 DNA ligase at a plasmid to fragment ratio of 1:2. All plasmids were confirmed by DNA sequencing.
Table 1.
List of primers used for site-directed mutagenesis and SOEing PCRa.
| Primer | Sequence (5′–3′) | |
|---|---|---|
| A | NCCR early SpeI | GGGGAAATCACTAGTCTTTTGCAATTTTTGCAAAAATGG |
| B | NCCR late SacII | GACAAGGCCAAGATTCCGCGGCTCGCAAAACATGTC |
| C | Tag PmlI | ACACCACCCCCAAAATAACACGTGCTTAAAAGTGGCTTATAC |
| D | NCCR bridge For | TAGTAAGGGTGTGGAAGCT |
| E | NCCR bridge Rev | CCATGGATTCTTCCCTGTTA |
| F | TCH1 SacII | GACAAGGCCAAGATTCCGCGGAGAATTTTAGGGGCGG |
| G | Dik3 C65T | GGCCTCAGAAAAAGCTTCCACACCCTTACTAC |
| H | TCH2 T65C | GGCCTCAGAAAAAGCCTCCACACCCTTACTAC |
Restriction enzyme sites are underlined and point mutations are shown in bold in the primer sequence.
Site-directed mutagenesis was performed on both Dik3 and the TCH2 clone to create and revert to O block polymorphism found in clinical variants, respectively. The C to T mutation was made in Dik3 using Primer G and its reverse complement and the T to C mutation was made in TCH2 using Primer H and its reverse complement (Table 1). Mutations were confirmed by DNA sequencing. The integrity of the early coding region was also confirmed by DNA sequencing.
BKV-NCCRs were amplified by PCR as previously described (Vanchiere et al., 2005b) from the urine of a patient with AIDS and BKV viruria. NCCR amplicons were cloned using the TOPO-TA system as directed by the manufacturer (Invitrogen, Carlsbad) and then into the Dun3 backbone. The Dun3 O block, containing the origin of replication and the SpeI site was added to each NCCR fragment by a Splicing by Overlap Extension (SOEing) PCR reaction (Horton et al., 1989). Additionally a SacII site was added to the existing S block of each NCCR by site directed mutagenesis. However, the SacII site was added into the truncated R block in the TCH1 clone because the S block is deleted in this clone. A bridge piece containing the O block and SpeI site was created by amplifying that region of the Dun3 plasmid using primers D and E. The bridge piece and the NCCR fragment were combined in a melting and slow cooling reaction using the program: 5 min at 95°C; 40 cycles, 30 sec each of a gradient from 95°C to 55°C; annealing at 68°C for 5 min. The forward SacII primer B, or F for clone TCH1, and the reverse SpeI primer A were then added to the above reaction at 4 nM each and PCR amplification was performed using the program: 3 min 30 sec at 94°C; 30 cycles of 45 sec at 94°C, 1 min at 66°C, 1 min at 68°C, annealing and extension at 68°C for 7 min. Fragments were then swapped into the Dun3 backbone as described above. GenBank accession numbers for the NCCRs are as follows: TCH1, HQ148166; TCH2, HQ148167; TCH3, HQ148168; TCH4, HQ148169; TCH5, HQ148170.
Transfections
The viral genome was excised from the pBR322 vector by BamHI (New England Biolabs, Ipswich, MA) digestion and then recirularized at a concentration of 10 ng/μL using T4 DNA ligase (New England Biolabs, Ipswich, MA). RPTE cells were seeded into 12 well plates and transfected with 0.6 μg DNA at 60–70% confluency using TransIT LT-1 transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer’s instructions with a DNA to transfection reagent ratio of 1:6. Transfection complexes were removed 18 h post-transfection by washing once with PBS and adding fresh media.
Southern Blotting
Low molecular weight DNA was harvested four or five days post-transfection (dpt) by the Hirt protocol (Hirt, 1967). DNA was linearized by digesting with BamHI or EcoRI and then digested with DpnI, which cuts only methylated DNA that has not been replicated in eukaryotic cells (Pipas et al., 1983). The sample was separated on a 1% agarose gel in 1X TAE buffer (40 mM Tris acetate and 1 mM EDTA, pH 7.8). The gel was washed with depurination solution (0.25 M HCl for 30 min), followed by denaturation solution (0.5 M NaOH, 1.5 M NaCl, twice for 20 min), followed by neutralization solution (0.5 M Tris-HCl, pH 7.4, 1.5 M NaCl, twice for 20 min). The gel was transferred overnight to nylon membrane (Gene Screen Plus, Perkin Elmer) in 20X SSC using capillary transfer. The membrane was washed for 5 min in 6X SSC and exposed to UV light for 2 min, then baked for 2 h at 80°C to fix the DNA to the membrane. The probe used was a 3239 bp BKV PvuII fragment excised from pGEM7-TU (another rearranged variant of BKV) which spans the early region and the NCCR, and was labeled with the Random Primers DNA Labeling System (Invitrogen). The membrane was rehydrated in 6X SSC and incubated for 1 h at 68°C in prehybridization solution (5X SSC, 1X Denhardt’s solution, 1% SDS) with 0.1 mg/mL denatured sheared salmon sperm DNA. The membrane was incubated overnight at 68°C in hybridization solution (5X SSC, 1X Denhardt’s solution, 1% SDS) with 0.1 mg/mL denatured sheared salmon sperm DNA and 1×108 counts per minute of labeled probe. After hybridization, the membrane was washed with 2X SSC with 0.1% SDS twice for 5 min at room temperature followed by 0.2X SSC with 0.1% SDS twice for 5 min at room temperature and twice for 15 min at 42°C. The membrane was then rinsed with 2X SSC before being exposed to film. The Southern blot was quantified using a Typhoon phosphoimager and Imagequant software (GE).
Acknowledgments
We thank Kathy Spindler, Christiane Wobus, Beth Moore, and members of the Imperiale lab for comments and discussion. This work was supported by NIH grant AI060584 awarded to M.J.I and in part by NIH grant CA046592 awarded to the University of Michigan Cancer Center and by NIH grant CA 104818 awarded to J.S.B. J.A.V was supported by NIH grant DK62090. N.M.B. was supported in part by the NIH National Research Service Award T32-GM07544 from the National Institute of General Medicine Sciences. J.R.A was supported by the NIH National Research Service Award T32-GM07544 and by the Frederick G. Novy Fellowship. S.M.B was supported by the Mary Sue and Kenneth Coleman Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nicole M. Broekema, Email: broekenm@umich.edu.
Shauna M. Bennett, Email: shaunamb@umich.edu.
Janet S. Butel, Email: jbutel@bcm.edu.
John A. Vanchiere, Email: jvanch@lsuhsc.edu.
Michael J. Imperiale, Email: imperial@umich.edu.
References
- Abend JR, Imperiale MJ. Transforming growth factor-beta-mediated regulation of BK virus gene expression. Virology. 2008;378:6–12. doi: 10.1016/j.virol.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abend JR, Low JA, Imperiale MJ. Inhibitory effect of gamma interferon on BK virus gene expression and replication. J Virol. 2007;81:272–9. doi: 10.1128/JVI.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesters PM, Heritage J, McCance DJ. Persistence of DNA sequences of BK virus and JC virus in normal human tissues and in diseased tissues. J Infect Dis. 1983;147:676–84. doi: 10.1093/infdis/147.4.676. [DOI] [PubMed] [Google Scholar]
- Cubukcu-Dimopulo O, Greco A, Kumar A, Karluk D, Mittal K, Jagirdar J. BK virus infection in AIDS. Am J Surg Pathol. 2000;24:145–9. doi: 10.1097/00000478-200001000-00019. [DOI] [PubMed] [Google Scholar]
- de Villiers J, Schaffner W, Tyndall C, Lupton S, Kamen R. Polyoma virus DNA replication requires an enhancer. Nature. 1984;312:242–6. doi: 10.1038/312242a0. [DOI] [PubMed] [Google Scholar]
- Del Vecchio AM, Steinman RA, Ricciardi RP. An element of the BK virus enhancer required for DNA replication. J Virol. 1989;63:1514–24. doi: 10.1128/jvi.63.4.1514-1524.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerries K. Human polyomavirus JC and BK persistent infection. Adv Exp Med Biol. 2006;577:102–16. doi: 10.1007/0-387-32957-9_8. [DOI] [PubMed] [Google Scholar]
- Eichman BF, Fanning E. The power of pumping together; deconstructing the engine of a DNA replication machine. Cell. 2004;119:3–4. doi: 10.1016/j.cell.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet. 1971;1:1253–7. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- Gorrill T, Feliciano M, Mukerjee R, Sawaya BE, Khalili K, White MK. Activation of early gene transcription in polyomavirus BK by human immunodeficiency virus type 1 Tat. J Gen Virol. 2006;87:1557–66. doi: 10.1099/vir.0.81569-0. [DOI] [PubMed] [Google Scholar]
- Gosert R, Rinaldo CH, Funk GA, Egli A, Ramos E, Drachenberg CB, Hirsch HH. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J Exp Med. 2008;205:841–52. doi: 10.1084/jem.20072097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudsmit J, Baak ML, Sleterus KW, Van der Noordaa J. Human papovavirus isolated from urine of a child with acute tonsillitis. Br Med J (Clin Res Ed) 1981;283:1363–4. doi: 10.1136/bmj.283.6303.1363-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudsmit J, Wertheim-van Dillen P, van Strien A, van der Noordaa J. The role of BK virus in acute respiratory tract disease and the presence of BKV DNA in tonsils. J Med Virol. 1982;10:91–9. doi: 10.1002/jmv.1890100203. [DOI] [PubMed] [Google Scholar]
- Heritage J, Chesters PM, McCance DJ. The persistence of papovavirus BK DNA sequences in normal human renal tissue. J Med Virol. 1981;8:143–50. doi: 10.1002/jmv.1890080208. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–9. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–8. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Jeffers LK, Madden V, Webster-Cyriaque J. BK virus has tropism for human salivary gland cells in vitro: implications for transmission. Virology. 2009;394:183–93. doi: 10.1016/j.virol.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Jin L, Gibson PE, Knowles WA, Clewley JP. BK virus antigenic variants: sequence analysis within the capsid VP1 epitope. J Med Virol. 1993;39:50–6. doi: 10.1002/jmv.1890390110. [DOI] [PubMed] [Google Scholar]
- Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles WA. The epidemiology of BK virus and the occurence of antigenic and genomic subtypes. In: Khalili K, Stoner GL, editors. Human Polyomaviruses: Molecular and Clinical Perspectives. Wiley-Liss; New York: 2001. [Google Scholar]
- Knowles WA. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV) Adv Exp Med Biol. 2006;577:19–45. doi: 10.1007/0-387-32957-9_2. [DOI] [PubMed] [Google Scholar]
- Knowles WA, Pillay D, Johnson MA, Hand JF, Brown DW. Prevalence of long-term BK and JC excretion in HIV-infected adults and lack of correlation with serological markers. J Med Virol. 1999;59:474–9. [PubMed] [Google Scholar]
- Low J, Humes HD, Szczypka M, Imperiale M. BKV and SV40 infection of human kidney tubular epithelial cells in vitro. Virology. 2004;323:182–8. doi: 10.1016/j.virol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Low JA, Magnuson B, Tsai B, Imperiale MJ. Identification of gangliosides GD1b and GT1b as receptors for BK virus. J Virol. 2006;80:1361–6. doi: 10.1128/JVI.80.3.1361-1366.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz RB, Dynan WS. Binding of cellular proteins to the regulatory region of BK virus DNA. J Virol. 1988;62:3388–98. doi: 10.1128/jvi.62.9.3388-3398.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz RB, Thompson HC, Mueller JF, Cohen JA, Dynan WS. Incidence of BK virus and JC virus viruria in human immunodeficiency virus-infected and -uninfected subjects. J Infect Dis. 1993;167:13–20. doi: 10.1093/infdis/167.1.13. [DOI] [PubMed] [Google Scholar]
- McNees AL, White ZS, Zanwar P, Vilchez RA, Butel JS. Specific and quantitative detection of human polyomaviruses BKV, JCV, and SV40 by real time PCR. J Clin Virol. 2005;34:52–62. doi: 10.1016/j.jcv.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Moens U, Johansen T, Johnsen JI, Seternes OM, Traavik T. Noncoding control region of naturally occurring BK virus variants: sequence comparison and functional analysis. Virus Genes. 1995;10:261–75. doi: 10.1007/BF01701816. [DOI] [PubMed] [Google Scholar]
- Moens U, Rekvig OP. Molecular Biology of BK Virus and Clinical and Basic Aspects of BK Virus Renal Infection. In: Khalili K, Stoner GL, editors. Human Polyomavirus. Wiley-Liss; New York: 2001. [Google Scholar]
- Moens U, Van Ghelue M. Polymorphism in the genome of non-passaged human polyomavirus BK: implications for cell tropism and the pathological role of the virus. Virology. 2005;331:209–31. doi: 10.1016/j.virol.2004.10.021. [DOI] [PubMed] [Google Scholar]
- O’Connor DT, Subramani S. Do transcriptional enhancers also augment DNA replication? Nucleic Acids Res. 1988;16:11207–22. doi: 10.1093/nar/16.23.11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GH, Hirsch HH, Rinaldo CH. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J Med Virol. 2009;81:1959–67. doi: 10.1002/jmv.21605. [DOI] [PubMed] [Google Scholar]
- Pipas JM, Peden KW, Nathans D. Mutational analysis of simian virus 40 T antigen: isolation and characterization of mutants with deletions in the T-antigen gene. Mol Cell Biol. 1983;3:203–13. doi: 10.1128/mcb.3.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein R, Schoonakker BC, Harley EH. Recurring theme of changes in the transcriptional control region of BK virus during adaptation to cell culture. J Virol. 1991;65:1600–4. doi: 10.1128/jvi.65.3.1600-1604.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif I, Khoury G, Dhar R. The genome of human papovavirus BKV. Cell. 1979;18:963–77. doi: 10.1016/0092-8674(79)90209-5. [DOI] [PubMed] [Google Scholar]
- Smith RD, Galla JH, Skahan K, Anderson P, Linnemann CC, Jr, Ault GS, Ryschkewitsch CF, Stoner GL. Tubulointerstitial nephritis due to a mutant polyomavirus BK virus strain, BKV(Cin), causing end-stage renal disease. J Clin Microbiol. 1998;36:1660–5. doi: 10.1128/jcm.36.6.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WJ, Berger SL, Triezenberg SJ, Folk WR. Nucleotides in the polyomavirus enhancer that control viral transcription and DNA replication. Mol Cell Biol. 1987;7:1681–90. doi: 10.1128/mcb.7.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanchiere JA, Abudayyeh S, Copeland CM, Lu LB, Graham DY, Butel JS. Polyomavirus shedding in the stool of healthy adults. J Clin Microbiol. 2009;47:2388–91. doi: 10.1128/JCM.02472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanchiere JA, Nicome RK, Greer JM, Demmler GJ, Butel JS. Frequent detection of polyomaviruses in stool samples from hospitalized children. J Infect Dis. 2005a;192:658–64. doi: 10.1086/432076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanchiere JA, White ZS, Butel JS. Detection of BK virus and simian virus 40 in the urine of healthy children. J Med Virol. 2005b;75:447–54. doi: 10.1002/jmv.20287. [DOI] [PubMed] [Google Scholar]
- Wilderman PJ, Hu B, Woodworth ME. Conformational changes in simian virus 40 rearranged regulatory regions: effects of the 21-base-pair promoters and their location. J Virol. 1999;73:10254–63. doi: 10.1128/jvi.73.12.10254-10263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AS, Cheng VC, Yuen KY, Kwong YL, Leung AY. High frequency of polyoma BK virus shedding in the gastrointestinal tract after hematopoietic stem cell transplantation: a prospective and quantitative analysis. Bone Marrow Transplant. 2009;43:43–7. doi: 10.1038/bmt.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RC, Wu R. Comparative study of papovavirus DNA: BKV(MM), BKV(WT) and SV40. Nucleic Acids Res. 1979;7:651–68. doi: 10.1093/nar/7.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng HY, Nishimoto Y, Chen Q, Hasegawa M, Zhong S, Ikegaya H, Ohno N, Sugimoto C, Takasaka T, Kitamura T, Yogo Y. Relationships between BK virus lineages and human populations. Microbes Infect. 2007;9:204–13. doi: 10.1016/j.micinf.2006.11.008. [DOI] [PubMed] [Google Scholar]