

Abstract
The world is currently undergoing a pandemic caused by an H1N1 influenza A virus, the so-called ‘swine flu’. The H5N1 (‘bird flu’) influenza A viruses, now circulating in Asia, Africa and Europe, are extremely virulent in humans, although they have not so far acquired the ability to transfer efficiently from human to human. These health concerns have spurred considerable interest in understanding the molecular biology of influenza A viruses. Recent structural studies of influenza A virus proteins (or fragments) help enhance our understanding of the molecular mechanisms of the viral proteins and the effects of drug resistance to improve drug design. The structures of domains of the influenza RNA-dependent RNA polymerase and the nonstructural NS1A protein provide opportunities for targeting these proteins to inhibit viral replication.
Influenza A viruses are responsible for sporadic pandemics that usually cause higher mortality rates than seasonal influenza epidemics. The most severe pandemic occurred in 1918, resulting in approximately 40 million deaths worldwide1. There were also pandemics in 1957 and 1968. In fact, we are currently in the midst of a pandemic caused by a virus originating in swine, the 2009 H1N1 virus or ‘swine flu’2,3. In addition, H5N1 viruses (‘bird flu’), which are also currently circulating, are extremely virulent in humans but have not yet acquired the ability for efficient human-to-human transmission (http://www.who.int/csr/disease/avian_influenza/country/cases_table_2009_07_01/en/index.html).
Influenza A viruses infect a wide range of avian and mammalian hosts, unlike influenza B viruses, which infect only humans. The envelope of influenza A viruses contains two different surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA)4,5. Influenza A viruses are categorized into antigenic HA and NA subtypes: 16 HA (H1–H16) and 9 NA (N1–N9) antigenic subtypes have been identified so far. Swine flu is an H1N1 virus because it contains a H1 subtype HA and a N1 subtype NA. The major influenza A subtypes that have infected humans during seasonal epidemics are H1N1, H2N2 and H3N2. Within a subtype, different strains arise as a result of point mutations. Influenza A viruses evolve constantly, and new mutant strains replace the old, in the process known as ‘genetic drift’. Each year, new influenza vaccines are designed by predicting the genetic drift of seasonal influenza A. Usually a pandemic occurs because of the emergence of a virus containing a new HA subtype, such as the H3 subtype in the 1968 pandemic. New HA subtypes are derived from wild birds, which are the reservoir of influenza A viruses, and are subsequently incorporated into human virus strains via reassortment of genomic RNA segments between human and avian influenza A virus strains. Alternatively, all eight genomic RNAs may be derived from an avian virus, and such a progenitor virus then undergoes multiple mutations in the process of adapting to the mammalian host6.
The current swine flu has emerged from reassortment of gene segments from North American and Eurasian swine strains that have been undetectably circulating in humans for a long period of time3. The H1-subtype HA of swine flu differs substantially from recent H1 HAs of seasonal influenza A viruses. Consequently, most of the human population lacks immunological protection against this virus, resulting in a pandemic. It is unusual for a pandemic virus to have the same HA subtype as currently circulating seasonal strains.
The primary defense against influenza A has been vaccination with inactivated or live-attenuated virus that is reformulated each year. Antivirals have also been used for both prophylactic and therapeutic treatments during seasonal epidemics5. Additionally, antivirals are particularly important at the beginning of a fast-spreading pandemic because the timely production of sufficient amounts of an effective vaccine is difficult. Current antivirals are directed against the M2 ion-channel protein (adamantanes) and NA (zanamivir and oseltamivir). However, many influenza A virus strains7 have developed resistance to adamantanes and/or oseltamivir (http://www.who.int/csr/disease/influenza/h1n1_table/en/index.html), highlighting the need for the development of new antiviral drugs. These health concerns have spurred considerable interest in understanding the molecular biology of influenza A viruses, and numerous structural studies of influenza virus proteins have been published in the last few years. These studies, in combination with extensive molecular biology and virology research initiatives, have provided opportunities to investigate several viral proteins and their interactions as targets for new chemotherapeutic agents.
Influenza A contains eight negative-stranded RNA genomic segments. The three largest RNA segments encode the three viral RNA-dependent RNA polymerase (RdRP) proteins: polymerase acidic protein (PA), polymerase basic protein 1 (PB1) and PB2. The RNA segment for PB1 also encodes a small 87-residue nonstructural protein, PB1-F2, which has apoptotic functions8. The three intermediate-size RNA segments encode HA, NA and the nucleoprotein. The larger of the remaining two segments encodes the M1 matrix protein and the M2 ion-channel protein, and the smaller one encodes two nonstructural proteins, NS1A and NS2/NEP. Here we present a structural-biology perspective on the existing and emerging molecular targets for anti-influenza drugs. The viral proteins are discussed in the order of their primary functions in the influenza A life cycle as outlined in the schematic representation in Figure 1.
Figure 1.

Influenza A life cycle. (a) Influenza A virus has a lipid bilayer envelope, within which are eight RNA genomic segments, each of which is associated with the trimeric viral RNA polymerase (PB1, PB2, PA) and coated with multiple nucleoproteins (NPs) to form the vRNPs. The outer layer of the lipid envelope is spiked with multiple copies of HA, NA and a small number of M2, whereas the M1 molecules keep vRNPs attached to the inner layer. (b) The viral surface glycoprotein HA binds to the host cell-surface sialic acid receptors, and the virus is transported into the cell in an endocytic vesicle. The low pH in the endosome triggers a conformational change in the HA protein that leads to fusion of the viral and endosomal membranes. The low pH also triggers the flow of protons into the virus via the M2 ion channel, thereby dissociating the vRNPs from M1 matrix proteins. The vRNPs that are released into the cytoplasm are transported into the nucleus by recognition of the nuclear localization sequences (NLSs) on nucleoproteins83 only when the M1 molecules are dissociated. (c) In the nucleus, the viral polymerase initiates viral mRNA synthesis with 5′-capped RNA fragments cleaved from host pre-mRNAs. The PB2 subunit binds the 5′ cap of host pre-mRNAs84, and the endonuclease domain in PA subunit cleaves the pre-mRNA 10–13 nucleotides downstream from the cap85. Viral mRNA transcription is subsequently initiated from the cleaved 3′ end of the capped RNA segment85,86. This ‘cap snatching’ occurs on nascent pre-mRNAs. (d) Viral mRNAs are transported to the cytoplasm for translation into viral proteins. The surface proteins HA, M2 and NA are processed in the endoplasmic reticulum (ER), glycosylated in the Golgi apparatus and transported to the cell membrane. (e) The NS1 protein of influenza A virus serves a critical role in suppressing the production of host mRNAs by inhibiting the 3′-end processing of host pre-mRNAs60,87, consequently blocking the production of host mRNAs, including interferon-β mRNAs. Unlike host pre-mRNAs, the viral mRNAs do not require 3′-end processing by the host cell machinery. Therefore, the viral mRNAs are transported to the cytoplasm, whereas the host mRNA synthesis is predominantly blocked. (f) The viral polymerase is responsible for not only capped RNA-primed mRNA synthesis but also unprimed replication of vRNAs in steps (−) vRNA → (+) cRNA → (−) vRNA. The nucleoprotein molecules are required for these two steps of replication and are deposited on the cRNA and vRNA during RNA synthesis30. The resulting vRNPs are subsequently transported to the cytoplasm, mediated by a M1–NS2 complex that is bound to the vRNPs; NS2 interacts with human CRM1 protein that exports the vRNPs from the nucleus88. (g) The vRNPs reach the cell membrane to be incorporated into new viruses (reviewed in ref. 89) that are budded out. The HA and NA proteins in new viruses contain terminal sialic acids that would cause the viruses to clump together and adhere to the cell surface. The NA of newly formed viruses cleaves these sialic acid residues, thereby releasing the virus from the host cell.
Hemagglutinin
HA molecules, which form trimers, attach the virus to sialic acid receptors on the cell surface and mediate the release of viral ribonucleoprotein particles (vRNPs) into the cytoplasm. A newly synthesized ~70-kDa HA is cleaved into HA1 and HA2, which are disulfide linked (Fig. 2a). HA1 contains the sialic acid binding site. After binding, the virus is internalized in endosomes (Fig. 2b). Acidification of the endosomes triggers a marked, irreversible conformational change in HA. This change includes dissociation of HA1 from the endosomal membrane and its movement away from HA2; a loop-to-helix transition in HA2 enables the fusion peptide at the N terminus of HA2 to attach to the endosomal membrane (see refs. 9,10 for details) and promote the fusion of the viral and endosomal membranes, resulting in the release of the vRNPs into the cytoplasm.
Figure 2.

Structural arrangement of HA trimers at prefusion state. (a) The HA precursor (HA0) is cleaved into sialic acid receptor binding domain (HA1) and ectodomain (HA2) that remain disulfide linked; TM, transmembrane anchor. (b) HA (HA1 + HA2) exists in trimeric form with a combined mass of ~220 kDa. Two HA molecules in a trimer90 are represented by their combined molecular surface, whereas the third one is shown in a ribbon representation, color coded according to panel a. The head of the HA1 chain (blue), which binds sialic acid receptors, has essentially all the antigenic sites against which human antibodies are directed. The HA1 head region is closely associated with the central coiled-coil trimeric α-helices (green). At fusion (low) pH, all three HA1 heads swing away from HA2 (ref. 91). The loops (red) connecting the coiled-coil stem to the fusion peptides are refolded to form helical structures that extend the coiled coil and expose the fusion peptides at the N-terminal end of HA2 (NHA2), which then contacts the endosome membrane for the fusion step9. Binding of the small-molecule inhibitor TBHQ12 (orange) at the HA:HA interface or binding of human monoclonal antibody CR6261 (yellow)13 or F10 (salmon)14 to a conserved region adjacent to the TBHQ pocket inhibits the low pH conformational change of HA.
Small molecules that block this irreversible reorganization of HA would be expected to inhibit virus entry. tert-Butylhydroquinone (TBHQ)11 binds prefusion HA trimers (Fig. 2b) at the interface between HA monomers12 and inhibits the low-pH conformational change of HA. The way TBHQ fits into this pocket suggests that specific modifications of this inhibitor would improve binding affinity, possibly leading to an antiviral drug. The TBHQ binding site is present only in group 2 HAs, which includes the H3 HA subtype, and is absent in group 1 HAs, which includes the subtypes H1, H2 and H5.
Although the vast majority of human antibodies are directed against several regions of the head of the HA1 (Fig. 2b), which undergoes frequent antigenic change, two human monoclonal antibodies, CR6261 (ref. 13) and F10 (ref. 14), have been shown to neutralize diverse influenza A subtypes. Structural studies of these human monoclonal antibodies in complex with HA have identified a conserved epitope near the base of HA to which the heavy chain of either antibody binds (Fig. 2b). This region is near the TBHQ binding pocket, indicating that binding of these antibodies would inhibit the low-pH conformational change of HA. Because administering F10 to mice protects against subsequent influenza A virus infection14, it is conceivable that these monoclonal antibodies might be used for passive immunity in humans.
M2 ion-channel protein
The M2 protein, specific to influenza A viruses, is the target for the adamantane drugs amantadine and rimantadine (Fig. 3). The structure and function of M2 protein have been characterized using extensive biochemical, biophysical and structural studies; however, many aspects of the molecular basis of selective proton transport at low pH, drug binding, mechanism of inhibition and effects of drug-resistance mutations remain unclear15,16. The 97-residue M2 protein has a transmembrane (TM) domain and a C-terminal cytoplasmic amphiphilic helix. The proton channel features a tetrameric arrangement of TM helices (Fig. 3). A number of recent X-ray crystallographic as well as solution- and solid-state NMR structural studies of M2 TM domains under various conditions, including various M2 constructs with or without a cytoplasmic tail, different pH and with or without adamantanes, have shown significant conformational heterogeneity and malleability of the TM tetramer17-20. However, the conformational variations in the arrangements of TM helices might be influenced by protein construct lengths and experimental conditions. Highly conserved residues His37 and Trp41 are located in the proton channel and are critical in the proton transport process. His37 is protonated at low pH21, which enhances the proton flow22. Trp41 residues, positioned adjacent to His37, are clustered at high pH, forming a ‘channel gate’ that blocks the proton channel. The gate opens up at low pH in association with the rearrangement of TM helices18,23.
Figure 3.

Structure, function and inhibition of the proton channel M2 protein of influenza A. (a) The vRNPs are attached to the lipid bilayer membrane via M1 matrix proteins. Influx of the protons from endosome to virus through M2 channels releases vRNPs. (b) The adamantanes (amantadine and rimantadine) inhibit the proton flow through the tetrameric M2 channel. (c) X-ray and NMR structures of M2 channel. (i) crystal structure of the transmembrane (TM) domain of M2–amantadine complex at pH 5.3 in which the drug (orange) binds M2 near Ser31 (ref. 17); (ii) solution NMR structure of M2–rimantadine complex at pH 7.5 reveals that the drug binds the individual M2 TM helix near Trp41 (ref. 18) (this structure also contains the C terminus cytoplasmic tail helix, not shown in the figure); (iii) solid-state NMR structure of M2–amantadine complex at pH 7.5 (ref. 19) reveals the drug binding to the proton channel, which is analogous to that in structure i (however, the spatial arrangements of the TM helices are different); and (iv) the arrangement of M2 TM helices as revealed by a solid-state NMR study of amantadine-bound M2 TM helix at pH 8.8 (ref. 92). The side chains of Ser31 (gray), His37 (cyan) and Trp41 (yellow) are shown in each structure.
The most prevalent adamantane-resistance M2 mutation, S31N, is located along the inside rim of the pore (Fig. 3c). The exact site of adamantane drug binding to the M2 TM domain remains controversial. X-ray17 and solid-state NMR19 structures indicate that an amantadine molecule binds the proton channel near the Ser31 cluster (Fig. 3c, i and iii), thereby physically obstructing the pore (the so-called ‘pore-blocking model’). Moreover, solid-state NMR data show substantial chemical-shift perturbation for Ser31 upon binding of amantadine19, and a chimeric proton channel comprised of the adamantane-insensitive influenza B M2 protein and a stretch of pore residues from influenza A M2 becomes sensitive to amantadine24. Alternatively, the solution NMR structures of M2 bound to rimantadine suggested an allosteric mechanism of M2 inhibition18 (Fig. 3c, ii). Here rimantadine molecules bind in the vicinity of the Trp41 gate to four equivalent lipid-facing pockets formed by adjacent helices, locking the tetrameric TM domain in its closed state. A subsequent structure of the S31N mutant M2 and biochemical analyses proposed that the drug-resistance mutation impairs the binding of rimantadine to the allosteric lipid-facing pocket by destabilizing the closely packed TM helices20. From a drug-discovery point of view, new M2 inhibitors have been reported25; however, their effectiveness against adamantane-resistant viruses remains to be established.
Nucleoprotein
Nucleoprotein molecules encapsidate the viral single-stranded RNAs (ssRNAs); multiple copies of nucleoprotein molecules, a ssRNA genome segment and a polymerase complex (P complex) are packaged into each vRNP that is incorporated into the virus (Fig. 1). Nucleoprotein molecules also participate in the nuclear import and export of vRNPs and viral replication, and they interact with host proteins (see ref. 26). Structurally, nucleoprotein molecules are characterized as parts of vRNPs by electron microscopy27 or as isolated nucleoprotein molecules by X-ray crystallography28,29. A nucleoprotein molecule folds into a crescent shape with a head and a body domain (Fig. 4), and the ssRNA binding groove is located on the exterior surface between the two domains of the nucleoprotein. Each nucleoprotein molecule has a tail loop that is inserted into a neighboring nucleoprotein molecule, resulting in the oligomerization of nucleoprotein, which is required for vRNP formation. A 12-residue tip of this loop (residues 408–419) is tightly gripped in a 16 × 16 × 10 Å3 loop-binding cavity between the head and body domains at the back of a neighboring nucleoprotein molecule. This interface involves both hydrophilic and hydrophobic interactions. In particular, a deeply buried intersubunit salt bridge between Arg416 in the tail loop and Glu339 in the neighboring nucleoprotein molecule is critical in this nucleoprotein-nucleoprotein interaction, as established by site-directed mutagenesis experiments. Hydrophobic patches in the cavity interact with the hydrophobic side chains of Ile408, Pro410, Phe412, Val414 and Pro419 in the tail loop (Fig. 4). The loop-binding cavity of nucleoprotein may be a viable target for small-molecule drugs that mimic the key structural features of the tail loop and would be expected to interfere with the oligomerization of nucleoprotein molecules. Nucleoprotein is also required for viral RNA replication, and recent evidence shows that the interaction of nucleoprotein with the viral polymerase mediates the switch from capped–primed viral mRNA synthesis to unprimed viral RNA replication30. Identification and structural characterization of this nucleoprotein-polymerase interaction might also reveal another drug target.
Figure 4.
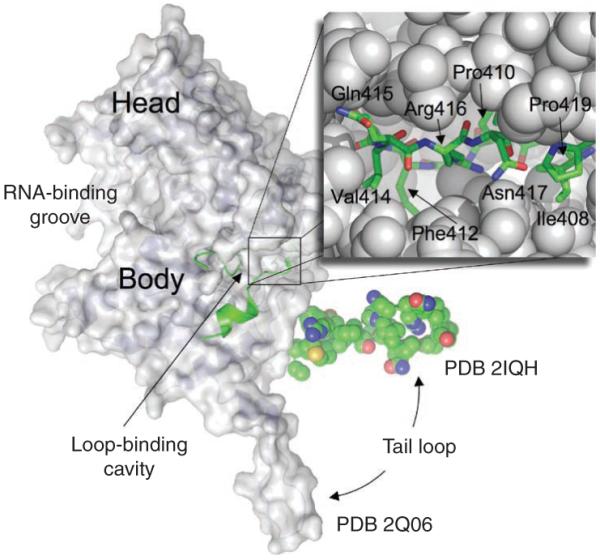
Structure of nucleoprotein. A nucleoprotein monomer has a crescent-shaped body and a tail loop. Comparison of two available crystal structures of nucleoprotein28,29 (PDB 2IQH and 2Q06) indicates the repositioning of the tail loop; the domain names28 are swapped to represent the current orientation. The tail loop of the neighboring nucleoprotein molecule (green) binds to a cavity at the back of the body domain. A zoomed view showing the binding of the tip of the tail loop (residues 408–419) to the loop-binding cavity, a potential site for drug design; the residues of the loop are labeled.
Viral polymerase
The viral polymerase (P complex) is a heterotrimer of subunits PA, PB1 and PB2 with a combined mass of ~250 kDa (Fig. 5a). P complex carries out both mRNA transcription (Fig. 1c) and replication (Fig. 1f). Transcription involves (i) binding of the 5′ cap (m7GTP) of a host pre-mRNA to the PB2 subunit (PB2cap; residues 318–482), (ii) cleaving of a phosphodiester bond 10–13 nucleotides downstream of the cap and (iii) initiating transcription of viral mRNAs at the cleaved 3′ end of the capped segment. The P complex also replicates the viral RNAs in a distinctly different process of unprimed initiation that requires nucleoprotein molecules. P complexes exist as heterotrimers or as a part of vRNPs, as revealed by electron microscopy studies27,31,32. However, the molecular mechanisms by which the P complex carries out the two distinct processes, transcription and replication, are poorly understood. Extensive biochemical and virological as well as recent structural studies have begun to reveal the architecture and specific roles of this complex.
Figure 5.

Structurally characterized fragments of influenza A polymerase complex (P-complex) that is involved in transcription and replication. (a) The heterotrimetric polymerase has subunits PA, PB1 and PB2 with a combined mass of ~250 kDa. The segments that have structural information are represented as colored bars—the colors correspond to the respective structures in the surrounding panels. (b) Ribbon representation of PAN domain (above) and electrostatic potential surface (below) of the endonuclease active site region35,36. We have docked an influenza endonuclease inhibitor, 2,4-dioxo-4-phenylbutanoic acid40 (yellow), that coordinates with the active-site metal ions (M1 and M2). The large cavity to which the phenyl ring points is a potential target for designing inhibitors with high binding affinity. (c) Ribbon representation (above) of PAC domain (green) in complex with an N-terminal helix of PB1N (gold) and the molecular surface of a section of the structure37,38 (below). The PB1N helix (P5TLLFLK11), shown in space-filling model, occupies a pocket which is a likely target for small-molecule inhibitors of PAC:PB1N dimerization. The PB1N helix has both hydrophobic and hydrogen-bond interactions with the PAC pocket residues that can be exploited for the inhibitor design. (d) Structure of PB1C–PB2N complex shows a ‘revolver-shaped’ helix bundle43. (e) The structure of the CAP (m7GTP) binding moiety PB2cap in complex with a m7GTP molecule45. (f) C-terminal region of PB2 contains a bipartite nuclear localized sequence (NLS) domain (blue), the structure of which was determined in complex with importin α5 (ref. 48). The adjacent domain (cyan) contains an RNA-binding cleft50.
The PA subunit (i) has endonuclease and protease activities33, (ii) is involved in vRNA/complementary RNA (cRNA) promoter binding34 and (iii) interacts with the PB1 subunit. Recently, crystal structures of the N- and C-terminal domains of PA (PAN, residues 1–197 (refs. 35,36), and PAC, residues 239–716 (refs. 37,38), respectively) in complex with the N-terminal domain of PB1 (PB1N) have been solved. The PAN domain (Fig. 5b) has a cation-dependent endonuclease active-site core39; the catalytic residues His41, Glu80, Asp108 and Glu119 are conserved among influenza A subtypes and strains. As discussed above, cleavage of host pre-mRNA by PAN is essential for the synthesis of viral mRNAs. A deep cleft at the endonuclease active site (Fig. 5b) of PAN35,36 is a promising site for structure-based design of novel anti-influenza drugs. Past efforts40,41 have generated influenza endonuclease inhibitors using structure-activity relationships (SAR) based on the concept of a common metal-dependent endonucleolytic processing39, as no PAN structure was available. Such an inhibitor from Merck, L-735882, had shown influenza endonuclease activity-specific inhibition with IC50 ~ 1 μM, and it did not inhibit host endonuclease, polymerase, RNase A or RNase H activities40. Recent crystal structures may enable structure-based design of other such inhibitors with improved potency and specificity. Our modeling of an inhibitor, 2,4-dioxo-4-phenylbutanoic acid (unpublished data), at the endonuclease active site (Fig. 5b) indicates that it would coordinate with the catalytic metal ions, and the phenyl ring would be positioned in an extended cavity. In agreement with published SAR data40, the docked model suggests that favorable substations at the phenyl ring with larger chemical groups would enhance inhibitor-protein interactions, whereas substitutions at the dioxobutanoic acid part could cause steric hindrance and/or eliminate metal coordination.
The PAC domain interacts with the N terminus of PB1N. The crystal structures of the PAC–PB1N complex37,38 (Fig. 5c) show that only the first 14 residues (2–15) of PB1N bind PAC; interactions between a small 310-helix carrying the residue sequence PTLLFLK of PB1N and a cleft flanked by four almost-parallel α-helices (α8, α10, α11 and α13) and a β-hairpin (β8–β9) are predominantly responsible for the stable complex formation between the domains of PA and PB1. The residues from PAC and PB1N at the interface are highly conserved in H1N1, H5N1 and other influenza A viruses. The PB1N binding core (Fig. 5c) in PAC, with an approximate height and diameter of about 15 and 10 Å, respectively, bears the chemical characteristics of a drug-binding site having potential to form four or five hydrogen bonds and elaborated hydrophobic interactions for binding small molecules. Mutations of key interface residues or use of a PB1N peptide inhibit viral replication and transcription38,42, which suggests a crucial role of the PAC-PB1N interactions in polymerase activity and/or heterotrimer formation. Therefore, novel chemotherapeutic agents mimicking the PB1N 310-helix are potential influenza inhibitors.
The PB1 subunit contains the RdRP active site and interacts with both PA and PB2 at distinct sites (Fig. 5a). A recently determined structure of a PB1C (residues 678–757)–PB2N (residues 1–35) complex43 reveals that three helices from each of the domains are bundled to form a ‘revolver-shaped’ structure (Fig. 5d). Mutations at the interface inhibit RNA synthesis, suggesting that compounds that can dissociate the PB1–PB2 complex would be potential influenza A inhibitors; however, a flat and extended interface may pose significant challenges in developing such a nonpeptide small-molecule inhibitor.
The PB2 subunit is responsible for cap binding44, and the C terminus of PB2 contains a bipartite nuclear localization signal (NLS) sequence for nuclear import from the cytoplasm. The crystal structure45 of the PB2 cap binding domain (PB2cap; residues 318–483) bound to a 5′-cap analog (m7GTP) (Fig. 5e) reveals a novel protein fold yet shares common features with known cap-binding proteins like eIF4E46 and vaccinia virus VP39 (ref. 47). In the PB2cap structure, the guanine base is sandwiched between His357 and Phe404, the guanine-base atoms N1 and N2 form a salt bridge with Glu361 and the presence of 7-methyl in the cap provides significant binding affinity for m7GTP versus GTP45. A cap mimic, if designed, would inhibit the transcription of influenza mRNAs; however, the similar cap-binding mode of host cap-binding proteins46 may pose a significant challenge for overcoming cytotoxicity of such an influenza inhibitor.
PB2C (residues 678–757; Fig. 5f) contains a bipartite NLS sequence that is responsible for nuclear import of the subdomain. A combined study of the solution NMR structure of PB2C and its crystal structure in complex with human importin α5 (ref. 48) showed the presence of a K736RKR739X(12)K752RIR bipartite NLS sequence49. An extended C-terminal fragment of PB2 (refs. 50,51) adjacent to the NLS-containing PB2C domain (Fig. 5f) has been structurally characterized and identified as an RNA binding domain (residues 535–684). The fragment contains the residue Lys627, which is important for viral replication in mammalian hosts; however, the absence of a complete structure of the P complex at atomic resolution fails to explain the relative arrangements of the functional domains and specific roles of residues such as Lys627 in viral replication. The replication of the viral RNA (vRNA→ cRNA → vRNA) by the influenza P complex seemingly shares a related structural architecture with the functionally comparable reovirus RNA polymerase λ3 (ref. 52). Based on superposition of the structures of PAC and the N-terminal domain of λ3 and docking of the PAC domain into the reconstructed electron microscopy structures of influenza polymerase complex, a previous study37 proposed a λ3 RdRP–based model53 for influenza polymerase. Structural characterization of the polymerase domain and biochemical information on the roles and interdependency of the domains of P complex may help reconstitute a more reliable atomic model of the complex. Nonetheless, the recent structures of domains of the P complex have started unraveling the promising target sites for designing new anti-influenza drugs.
Nonstructural protein NS1A
The multifunctional NS1A protein has two domains. NS1AN (residues 1–70) exists in a homodimeric state that binds double-stranded RNA (dsRNA) in a non–sequence specific manner. The structures of NS1AN54,55 alone and in complex with a dsRNA56 (Fig. 6a) revealed the dsRNA binding surface and mapped the specific RNA-protein interactions. A conserved residue, Arg38, from each of the NS1AN monomers is paired to interact with the dsRNA at its major grove56. The primary function of the NS1AN domain is the inhibition of the interferon (IFN)-α/β–induced 2′-5′-oligo A synthetase/RNase L pathway by sequestering dsRNA away from 2′-5′-oligo synthetase57. An influenza A/Udorn/307/72 virus (Udorn virus) that encodes an R38A mutant NS1A protein is highly attenuated57, indicating that drugs that interfere with NS1AN-RNA binding should inhibit virus replication. The NS1AN dimer has a small cavity at the center of its two-fold axis, and the cavity opens toward the RNA-binding surface. Structure-based design of compounds to specifically bind the cavity and interfere with the NS1AN-dsRNA binding may generate effective inhibitors of influenza A. Also, disruption of homodimer formation (Fig. 6a) would block dsRNA binding to NS1A.
Figure 6.
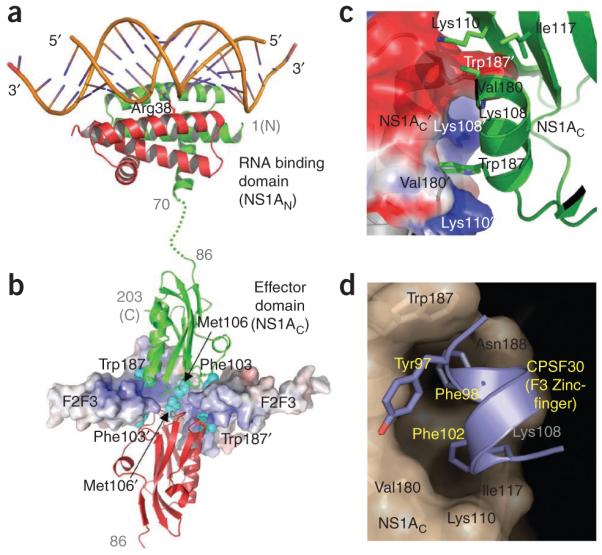
Structures of N- and C-terminal domains of influenza A nonstructural protein NS1A and complexes. (a) The N-terminal domain (NS1AN) forms a highly stable dimer that binds dsRNA in a non–sequence specific manner56. (b) The C-terminal effector domain (NS1AC) has been shown to bind F2F3 zinc fingers of human CPSF30 (ref. 62); this function of NS1A is essential to inhibit 3′-end processing of cellular pre-mRNAs60. (c) The dimer interface in apo NS1AC. (d) Upon binding of CPSF30, the NS1AC domain undergoes conformational changes (from c to d) to create a hydrophobic pocket that accommodates three aromatic residues of the F3 zinc finger. The pocket region is highly conserved in influenza A viruses and is a promising target for anti–influenza A drugs that would block a key host and viral protein interaction. In apo structures63,64, as represented in c, the pocket does not exist, and a hydrophobic patch at the region is hidden at an intermolecular interface, primarily due to the conserved Trp187 side chain.
NS1AC (residues 86–230/237), also known as the effector domain, binds several cellular proteins: (i) the p85β subunit of phosphatidylinositol-3 kinase (PI3K), leading to the activation of PI3K signaling58; (ii) protein kinase R (PKR), thereby inhibiting a PKR activation59 pathway that would otherwise cause the inhibition of protein synthesis and hence virus replication; and (iii) the 30-kDa subunit of cleavage and polyadenylation specificity factor (CPSF30) to inhibit 3′-end processing of cellular pre-mRNAs, including IFN-β pre-mRNA60. A fragment containing the second and third (F2F3) of the five zinc-binding domains of CPSF30 has been shown to inhibit influenza A viral replication and increase production of IFN-β mRNA61, showing that F2F3 blocks CPSF30 binding to NS1AC. A crystal structure of the NS1AC–F2F3 complex62 revealed the formation of a 2:2 tetrameric complex (Fig. 6b). Comparison of the structure of the complex with a structure of the apo NS1AC domain63 revealed significant structural rearrangement of NS1AC upon F2F3 binding: two NS1AC molecules in a head-to-head arrangement are wrapped with two F2F3 molecules and the apo NS1AC molecules are arranged side by side (Fig. 6c). The structure of the complex also revealed a hydrophobic CPSF30 binding pocket on the NS1AC surface (Fig. 6d) that is composed of highly conserved residues. A Udorn virus encoding NS1A with a single CPSF30 binding-pocket mutation (G184R) is attenuated and does not inhibit IFN-β pre-mRNA processing, confirming that the F2F3 fragment indeed blocks CPSF30 binding to NS1A. In the absence of F2F3 binding, the pocket (Fig. 6d) is not formed, and a key conserved surface hydrophobic residue Trp187 is hidden at a dimeric interface (Fig. 6c) between symmetry-related molecules in the apo structures63,64.
The hydrophobic CPSF30 binding pocket is conserved in almost all (>98%) influenza A viruses isolated from humans, except for residues Ile119 and Val180 at the edge of the pocket, which are substituted with hydrophobic homologs Met119 and Ile180, respectively, in some viruses. The NS1A proteins of several human influenza A viruses block IFN production via NS1A–CPSF30 binding65. Further evidence for the biological significance of the NS1A-CPSF30 complex and the common use of this pocket was revealed by the analysis of two residues in the NS1A protein, Phe103 and Met106, that are outside the conserved CPSF30 binding pocket (Fig. 6b). These two residues are involved in both NS1A-NS1A and NS1A-F2F3 interactions that stabilize the tetrameric NS1AC–F2F3 complex62. Phe103 and Met106 are highly conserved (99.7%) in the NS1A proteins of influenza A viruses isolated from humans. The NS1A protein of the 2009 H1N1 virus has the consensus CPSF30 binding sequence and consensus Phe103 and Met106 residues. The small number of human influenza A viruses that express a NS1A protein with residues other than Phe103 and Met106 includes the laboratory-generated influenza A/PR/8/34 strain65 and the pathogenic H5N1 viruses transmitted to humans in 1997 (ref. 61). Nonetheless, as shown by experiments using the H5N1 influenza A/Hong Kong/483/97 virus (HK97), the NS1A protein expressed by these 1997 H5N1 viruses, which contain Leu103 and Ile106, binds CPSF30 to a significant, though nonoptimal, extent in infected cells because its cognate viral polymerase complex (PB1, PB2, PA, nucleoprotein) stabilizes the NS1A–CPSF30 complex66. Because of weakened CPSF30 binding, the H5N1 HK97 virus is attenuated in tissue culture61 and shows decreased virulence in mice (M.J. Hossain, R.M.K. and R. Donis, unpublished data). In contrast, essentially all highly pathogenic H5N1 viruses in circulation since 2003 possess the consensus Phe103 and Met106 residues in NS1A and bind CPSF30 efficiently61.
The above facts establish that influenza A virus replication can be inhibited by interfering with NS1AC-CPSF30 binding, and the structural features of the F3 binding pocket on NS1AC show the characteristics of a viable drug target. Small molecules mimicking the three aromatic residues (Tyr97, Phe98 and Phe102) of F3 on a helix backbone are predicted to inhibit viral replication by inhibiting CPSF30 binding to NS1A62. Such an inhibitor will interfere with interactions between a viral and a host protein. This differs from the actions of all other proposed and existing influenza drugs that primarily inhibit viral enzymes or interfere with interactions between viral entities only.
Neuraminidase
The flu drugs oseltamivir67 and zanamivir68 are sialic acid–mimicking inhibitors of NA (Fig. 7a). These drugs were developed by structure-based drug-design efforts (see the review in ref. 69). The sialic acid binding pocket in NA was described when the initial crystal structure of NA was reported70,71. Chemical modifications and substitutions to a sialic acid backbone based on structural information led to the design of these drugs. For example, substituting the 4-hydroxyl group of sialic acid with a guanidine group in zanamivir (Fig. 7a,b) helped gain binding affinity through interactions with surrounding acidic-residue side chains of Asp151 and Glu227, main chain carbonyls of Asp151 and Trp178 and hydrophobic stacking with Glu119. The glycerol moiety, which is common to both sialic acid and zanamivir, has O-H…O type interactions with Glu276. Oseltamivir, which has the chemical substitutions of the glycerol part with a hydrophobic l-ethylpropoxy group and the guanidine group with an amine, reduced the polarity and increased lipophilicity of the compound, which could then be formulated as an oral pro-drug, in contrast to zanamivir, which is inhaled. A difference in oseltamivir binding compared with zanamivir and sialic acid binding is that Glu276 forms a hydrophobic stacking with the ethylpropoxy group of oseltamivir compared to its hydrogen bond with the glycerol moiety of zanamivir or sialic acid. Influenza viruses have developed oseltamivir resistance primarily through H274Y substitution in NA. A structural study revealed that the NA mutant72 discriminates against the l-ethylpropoxy group substitution in oseltamivir by repositioning the residue Glu276 through the H274Y mutation (Fig. 7b,c). The mutation R292K, which causes resistance to oseltamivir and a low level of resistance to zanamivir, also repositions the side chain of Glu276.
Figure 7.
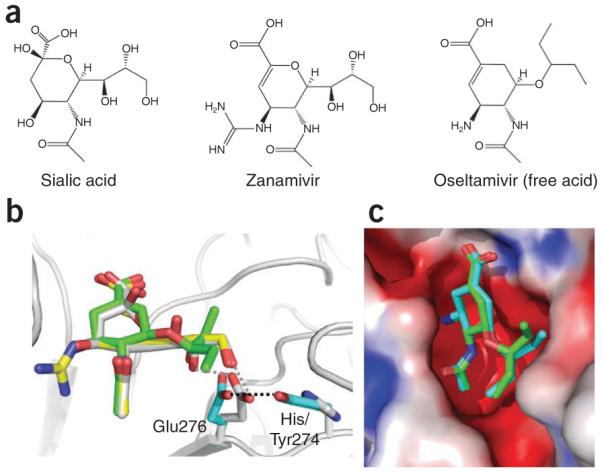
Binding of sialic acid–mimic drugs zanamivir and oseltamivir to neuraminidase (NA). (a,b) Chemical structures (a) and superposition of NA structures (b) show highly similar modes of binding for zanamivir (yellow) and oseltamivir (green) to sialic acid (gray) substrate, which reflects the influence of structures in the discovery of these drugs67,68. Recent crystal structures of oseltamivir-resistant mutant NA complexes72 show how the virus uses the NA H274Y mutation to reposition Glu276, which discriminates the l-ethylpropoxy group of oseltamivir from the glycerol moiety of the substrate. The repositioned Glu276 side chain would develop steric conflict with the l-ethylpropoxy group of oseltamivir, whereas it could still maintain favorable interactions with the glycerol part, common to both sialic acid and zanamivir. (c) Comparison of the modes of binding of oseltamivir to wild-type and H267Y mutant NA (shown in green and cyan, respectively). The H264Y mutation affects the positioning of the l-ethylpropoxy group of oseltamivir. The rearrangement of l-ethylpropoxy group of oseltamivir is associated with loss of inhibitor-protein interactions, resulting in a significant drug resistance.
A viable drug-resistant mutant discriminates against an inhibitor while retaining reasonable substrate-binding affinity. Therefore, an inhibitor that deviates least from the substrate would be least affected by resistance mutations; however, chemical modifications are often required to reduce cytotoxicity and improve pharmacokinetic characteristics of a drug candidate. Structures obtained from crystals of apo NA73 soaked with oseltamivir for a short period revealed the existence of a transient pocket adjacent to the sialic acid–binding pocket when compared to that observed in substrate- or inhibitor-bound NA structures. This new pocket, formed primarily by repositioning of the 150-loop73, suggests that NA undergoes conformational changes upon substrate binding. The adaptability of the region may be exploited to accommodate wider chemical substitutions in sialic acid mimics or allow the design of structurally distinct allosteric inhibitors to trap open conformations of the pocket region. Targeting the sialic acid binding pocket and the nearby pocket may resemble the ATP binding pocket and the adjacent pocket in protein kinases74. Extensive studies on targeting the ATP binding pocket and an adjacent non-ATP pocket in protein kinases may conceptually guide the design of new NA-inhibiting influenza drugs.
Conclusions
Treatment of seasonal and pandemic influenza is currently limited by the availability of only few drugs that are challenged by emergence of drug-resistant mutants. Analogous to the treatments used against HIV-1 infection, a combination of potent influenza drugs, each targeting a different viral entity or with different modes of inhibition, would be expected to be more effective in treating virulent and pandemic influenza A viruses. In fact, combined use of amantadine and oseltamivir has been shown to reduce the emergence of drug-resistant influenza A viruses75. Lessons learned from designing HIV-1 drugs, such as HIV-1 protease substrate mimics76 (the design strategy of which is related to that of sialic acid–mimic NA-inhibiting influenza drugs77) and allosteric non-nucleoside reverse transcriptase inhibitors78, may help in the development of influenza drugs against existing and emerging targets such that future influenza drugs will be more resilient to drug-resistant and pandemic influenza strains.
Note added in proof: the following relevant publications appeared while this review was in press: (i) a recent solid-state NMR spectroscopy study79 confirms the binding of amantadine to the pore (high-affinity binding site) and the allosteric lipid-facing pocket (low-affinity binding site); (ii) a crystal structure of NS1 effector domain in complex with the inter-SH2 (coiled-coil) domain of p85b subunit of PI3-K 80 suggests that the NS1:SH2 interactions, and possibly the NS1:p110 subunit interactions, are responsible for PI3-K activation; (iii) investigational drug T-705 (favipiravir), an inhibitor of viral RNA synthesis, is reported to have antiviral activity against oseltamivir-sensitive and oseltamivir-resistant H5N1 viruses81; and (iv) a new neuraminidase inhibitor is active against H5N1 and oseltamivir-resistant viruses82.
ACKNOWLEDGMENTS
We thank A.J. Shatkin, Y.J. Tao and G.T. Montelione for helpful discussions. J.M.A. and L.M. are supported by US National Institutes of Health grants U54GM074958 and U01AI74497, and R.M.K. is supported by US National Institutes of Health grants AI11772, AI074497 and U01AI174497 for conducting research on topics covered in this review.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Reid AH, Taubenberger JK, Fanning TG. The 1918 Spanish influenza: integrating history and biology. Microbes Infect. 2001;3:81–87. doi: 10.1016/s1286-4579(00)01351-4. [DOI] [PubMed] [Google Scholar]
- 2.Chowell G, et al. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N. Engl. J. Med. 2009;361:674–679. doi: 10.1056/NEJMoa0904023. [DOI] [PubMed] [Google Scholar]
- 3.Garten RJ, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamb RA, Krug RM. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4th edn. Lippincott, Williams & Wilkins; Philadelphia, USA: 2001. pp. 1487–1532. [Google Scholar]
- 5.Wright PF, Webster RG. Orthomyxoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 4th edn. Lippincott, Williams & Wilkins; Philadelphia, USA: 2001. pp. 1533–1579. [Google Scholar]
- 6.Taubenberger JK, et al. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- 7.Moscona A. Oseltamivir resistance—disabling our influenza defenses. N. Engl. J. Med. 2005;353:2633–2636. doi: 10.1056/NEJMp058291. [DOI] [PubMed] [Google Scholar]
- 8.Chen W, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 9.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 10.Harrison SC. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu KL, et al. Structure-activity relationships for a series of thiobenzamide influenza fusion inhibitors derived from 1,3,3-trimethyl-5-hydroxy-cyclohexylmethylamine. Bioorg. Med. Chem. Lett. 2002;12:3379–3382. doi: 10.1016/s0960-894x(02)00761-8. [DOI] [PubMed] [Google Scholar]
- 12.Russell RJ, et al. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA. 2008;105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cady SD, Luo W, Hu F, Hong M. Structure and function of the influenza A M2 proton channel. Biochemistry. 2009;48:7356–7364. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 17.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J. Mol. Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada A, Miura T, Takeuchi H. Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza a virus. Biochemistry. 2001;40:6053–6060. doi: 10.1021/bi0028441. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Lamb RA, Pinto LH. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys. J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 24.Jing X, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc. Natl. Acad. Sci. USA. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, et al. Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J. Am. Chem. Soc. 2009;131:8066–8076. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 27.Area E, et al. 3D structure of the influenza virus polymerase complex: localization of subunit domains. Proc. Natl. Acad. Sci. USA. 2004;101:308–313. doi: 10.1073/pnas.0307127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 29.Ng AK, et al. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008;22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newcomb LL, et al. Interaction of the influenza a virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J. Virol. 2009;83:29–36. doi: 10.1128/JVI.02293-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torreira E, et al. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res. 2007;35:3774–3783. doi: 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coloma R, et al. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009;5:e1000491. doi: 10.1371/journal.ppat.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanz-Ezquerro JJ, Zurcher T, de la Luna S, Ortin J, Nieto A. The amino-terminal one-third of the influenza virus PA protein is responsible for the induction of proteolysis. J. Virol. 1996;70:1905–1911. doi: 10.1128/jvi.70.3.1905-1911.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maier HJ, Kashiwagi T, Hara K, Brownlee GG. Differential role of the influenza A virus polymerase PA subunit for vRNA and cRNA promoter binding. Virology. 2008;370:194–204. doi: 10.1016/j.virol.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 35.Yuan P, et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 36.Dias A, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 37.He X, et al. Crystal structure of the polymerase PAC–PB1N complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 38.Obayashi E, et al. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature. 2008;454:1127–1131. doi: 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- 39.Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomassini J, et al. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994;38:2827–2837. doi: 10.1128/aac.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parkes KE, et al. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2003;46:1153–1164. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 42.Ghanem A, et al. Peptide-mediated interference with influenza A virus polymerase. J. Virol. 2007;81:7801–7804. doi: 10.1128/JVI.00724-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugiyama K, et al. Structural insight into the essential PB1–PB2 subunit contact of the influenza virus RNA polymerase. EMBO J. 2009;28:1803–1811. doi: 10.1038/emboj.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blaas D, Patzelt E, Keuchler E. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 1982;10:4803–4812. doi: 10.1093/nar/10.15.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guilligay D, et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 46.Marcotrigiano J, Gingras A-C, Sonenberg N, Burley SK. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell. 1997;89:951–961. doi: 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 47.Hodel AE, Gershon PD, Quiocho FA. Structural basis for sequence-nonspecific recognition of 5′-capped mRNA by a cap-modifying enzyme. Mol. Cell. 1998;1:443–447. doi: 10.1016/s1097-2765(00)80044-1. [DOI] [PubMed] [Google Scholar]
- 48.Tarendeau F, et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 2007;14:229–233. doi: 10.1038/nsmb1212. [DOI] [PubMed] [Google Scholar]
- 49.Fontes MR, Teh T, Jans D, Brinkworth RI, Kobe B. Structural basis for the specificity of bipartite nuclear localization sequence binding by importin-α. J. Biol. Chem. 2003;278:27981–27987. doi: 10.1074/jbc.M303275200. [DOI] [PubMed] [Google Scholar]
- 50.Kuzuhara T, et al. Structural basis of the influenza A virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J. Biol. Chem. 2009;284:6855–6860. doi: 10.1074/jbc.C800224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarendeau F, et al. Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog. 2008;4:e1000136. doi: 10.1371/journal.ppat.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shatkin AJ. Viruses with segmented ribonucleic acid genomes: multiplication of influenza versus reovirus. Bacteriol. Rev. 1971;35:250–266. doi: 10.1128/br.35.3.250-266.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage–structural studies of reovirus polymerase λ3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 54.Chien CY, et al. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol. 1997;4:891–895. doi: 10.1038/nsb1197-891. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, et al. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat. Struct. Biol. 1997;4:896–899. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]
- 56.Cheng A, Wong SM, Yuan YA. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2009;19:187–195. doi: 10.1038/cr.2008.288. [DOI] [PubMed] [Google Scholar]
- 57.Min J-Y, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ OAS/RNase L pathway. Proc. Natl. Acad. Sci. USA. 2006;103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hale BG, Jackson D, Chen YH, Lamb RA, Randall RE. Influenza A virus NS1 protein binds p85β and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA. 2006;103:14194–14199. doi: 10.1073/pnas.0606109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Min J-Y, Li S, Sen GC, Krug RM. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007;363:236–243. doi: 10.1016/j.virol.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 60.Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 61.Twu KY, Noah DL, Rao P, Kuo R-L, Krug RM. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 2006;80:3957–3965. doi: 10.1128/JVI.80.8.3957-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das K, et al. Structural basis for suppression by influenza A virus of a host antiviral response. Proc. Natl. Acad. Sci. USA. 2008;105:13093–13098. doi: 10.1073/pnas.0805213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bornholdt ZA, Prasad BV. X-ray structure of influenza virus NS1 effector domain. Nat. Struct. Mol. Biol. 2006;13:559–560. doi: 10.1038/nsmb1099. [DOI] [PubMed] [Google Scholar]
- 64.Bornholdt ZA, Prasad BV. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature. 2008;456:985–988. doi: 10.1038/nature07444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kochs G, Garcia-Sastre A, Martinez-Sobrido L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007;81:7011–7021. doi: 10.1128/JVI.02581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuo RL, Krug RM. Influenza a virus polymerase is an integral component of the CPSF30–NS1A protein complex in infected cells. J. Virol. 2009;83:1611–1616. doi: 10.1128/JVI.01491-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim CU, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis and structural analysis of carbocyclic sialiac acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 68.von Itzstein M, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 69.von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 70.Varghese JN, Laver WG, Colman PM. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- 71.Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303:41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- 72.Collins PJ, et al. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature. 2008;453:1258–1261. doi: 10.1038/nature06956. [DOI] [PubMed] [Google Scholar]
- 73.Russell RJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 74.Sebolt-Leopold JS, English JM. Mechanisms of drug inhibition of signalling molecules. Nature. 2006;441:457–462. doi: 10.1038/nature04874. [DOI] [PubMed] [Google Scholar]
- 75.Ilyushina NA, Bovin NV, Webster RG, Govorkova EA. Combination chemotherapy, a potential strategy for reducing the emergence of drug-resistant influenza A variants. Antiviral Res. 2006;70:121–131. doi: 10.1016/j.antiviral.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 76.Chellappan S, Kairys V, Fernandes MX, Schiffer C, Gilson MK. Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins. 2007;68:561–567. doi: 10.1002/prot.21431. [DOI] [PubMed] [Google Scholar]
- 77.Colman PM. New antivirals and drug resistance. Annu. Rev. Biochem. 2009;78:95–118. doi: 10.1146/annurev.biochem.78.082207.084029. [DOI] [PubMed] [Google Scholar]
- 78.Das K, Lewi PJ, Hughes SH, Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 79.Cady SD, et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hale BG, et al. Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein. Proc. Natl. Acad. Sci. USA. 2010;107:1954–1959. doi: 10.1073/pnas.0910715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kiso M, et al. T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc. Natl. Acad. Sci. USA. 2010;107:882–887. doi: 10.1073/pnas.0909603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kiso M, et al. Efficiency of the new neuraminidase inhibitor CS-8958 against H5N1 influenza viruses. PloS Pathogen. 2010;6:e10000786. doi: 10.1371/journal.ppat.1000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu WW, Pante N. The directionality of the nuclear transport of the influenza A genome is driven by selective exposure of nuclear localization sequences on nucleoprotein. Virol. J. 2009;6:68. doi: 10.1186/1743-422X-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ulmanen I, Broni BA, Krug RM. The role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. USA. 1981;78:7355–7359. doi: 10.1073/pnas.78.12.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonucleaase cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- 86.Hagen M, Chung TDY, Butcher A, Krystal M. Recombinant influenza virus polymerase: requirement of both 5′ and 3′ viral ends for endonuclease activity. J. Virol. 1994;68:1509–1515. doi: 10.1128/jvi.68.3.1509-1515.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shimizu K, Iguchi A, Gomyou R, Ono Y. Influenza virus inhibits cleavage of the HSP70 pre-mRNAs at the polyadenylation site. Virology. 1999;254:213–219. doi: 10.1006/viro.1998.9555. [DOI] [PubMed] [Google Scholar]
- 88.Neumann G, Hughes MT, Kawaoka Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000;19:6751–6758. doi: 10.1093/emboj/19.24.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nayak DP, Hui EK, Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106:147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 91.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 92.Hu J, et al. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys. J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]