

Abstract
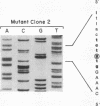
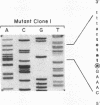
Human trifunctional protein catalyzes three steps in mitochondrial beta-oxidation of fatty acids, including the long chain 3-hydroxyacyl-CoA dehydrogenase step. Deficiency of this heterocomplex, which contains 4 alpha and 4 beta subunits, causes sudden unexplained infant death, a Reye-like syndrome, cardiomyopathy, or skeletal myopathy. We determined the molecular basis of this deficiency in a patient with neonatal presentation and later sudden death using reverse transcription and PCR amplification of his alpha subunit mRNA. We demonstrated a universal deletion of exon 3 (71 bp) in his mRNA. This deletion causes a frameshift and very early premature termination. Amplification of genomic DNA demonstrated that the patient was a compound heterozygote with two different mutations in the 5' donor splice site following exon 3: a paternally inherited G to A transversion at the invariant position +1 and a maternally inherited A to G mutation at position +3. Both allelic mutations apparently cause exon 3 skipping, resulting in undetectable levels of alpha subunit protein, and complete loss of trifunctional protein. This is the initial molecular characterization of trifunctional protein deficiency.
Full text
PDF



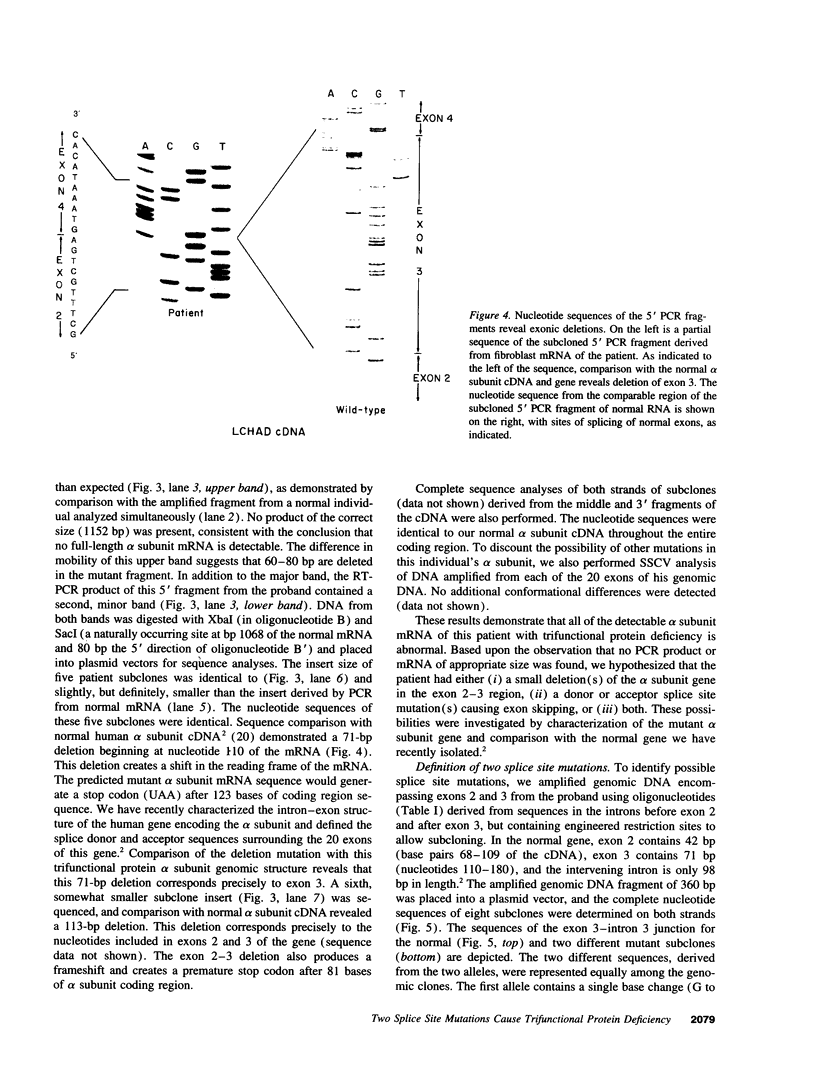
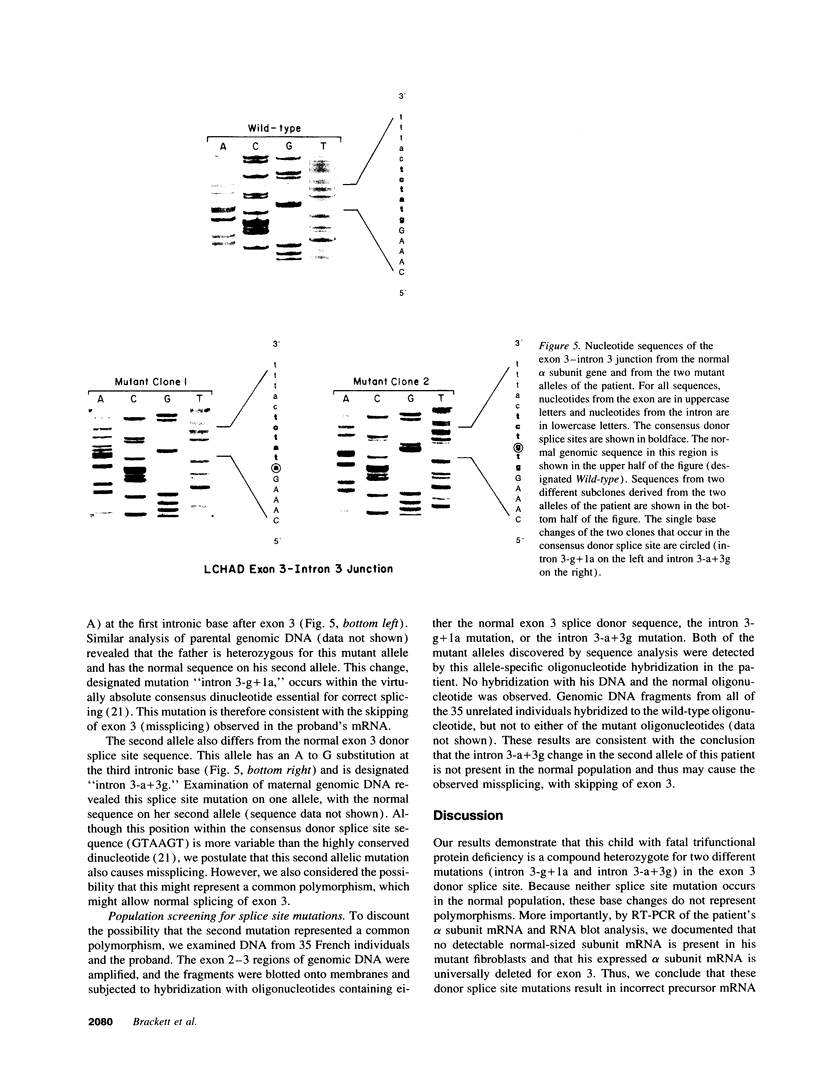


Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Brackett J. C., Sims H. F., Steiner R. D., Nunge M., Zimmerman E. M., deMartinville B., Rinaldo P., Slaugh R., Strauss A. W. A novel mutation in medium chain acyl-CoA dehydrogenase causes sudden neonatal death. J Clin Invest. 1994 Oct;94(4):1477–1483. doi: 10.1172/JCI117486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter K., Pollitt R. J., Middleton B. Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem Biophys Res Commun. 1992 Mar 16;183(2):443–448. doi: 10.1016/0006-291x(92)90501-b. [DOI] [PubMed] [Google Scholar]
- Carstens R. P., Fenton W. A., Rosenberg L. R. Identification of RNA splicing errors resulting in human ornithine transcarbamylase deficiency. Am J Hum Genet. 1991 Jun;48(6):1105–1114. [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987 Apr;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Frerman F. E., Goodman S. I. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci U S A. 1985 Jul;82(13):4517–4520. doi: 10.1073/pnas.82.13.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher P. G., Tse W. T., Costa F., Scarpa A., Boivin P., Delaunay J., Forget B. G. A splice site mutation of the beta-spectrin gene causing exon skipping in hereditary elliptocytosis associated with a truncated beta-spectrin chain. J Biol Chem. 1991 Aug 15;266(23):15154–15159. [PubMed] [Google Scholar]
- Ganguly A., Baldwin C. T., Strobel D., Conway D., Horton W., Prockop D. J. Heterozygous mutation in the G+5 position of intron 33 of the pro-alpha 2(I) gene (COL1A2) that causes aberrant RNA splicing and lethal osteogenesis imperfecta. Use of carbodiimide methods that decrease the extent of DNA sequencing necessary to define an unusual mutation. J Biol Chem. 1991 Jun 25;266(18):12035–12040. [PubMed] [Google Scholar]
- Garbarz M., Tse W. T., Gallagher P. G., Picat C., Lecomte M. C., Galibert F., Dhermy D., Forget B. G. Spectrin Rouen (beta 220-218), a novel shortened beta-chain variant in a kindred with hereditary elliptocytosis. Characterization of the molecular defect as exon skipping due to a splice site mutation. J Clin Invest. 1991 Jul;88(1):76–81. doi: 10.1172/JCI115307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoda T., Yamada N., Murase T., Inaba T., Ishibashi S., Shimano H., Koga S., Yazaki Y., Furuichi Y., Takaku F. Occurrence of multiple aberrantly spliced mRNAs upon a donor splice site mutation that causes familial lipoprotein lipase deficiency. J Biol Chem. 1991 Dec 25;266(36):24757–24762. [PubMed] [Google Scholar]
- Hale D. E., Bennett M. J. Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr. 1992 Jul;121(1):1–11. doi: 10.1016/s0022-3476(05)82532-6. [DOI] [PubMed] [Google Scholar]
- Jackson S., Bartlett K., Land J., Moxon E. R., Pollitt R. J., Leonard J. V., Turnbull D. M. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Res. 1991 Apr;29(4 Pt 1):406–411. doi: 10.1203/00006450-199104000-00016. [DOI] [PubMed] [Google Scholar]
- Jackson S., Kler R. S., Bartlett K., Briggs H., Bindoff L. A., Pourfarzam M., Gardner-Medwin D., Turnbull D. M. Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest. 1992 Oct;90(4):1219–1225. doi: 10.1172/JCI115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T., Aoyama T., Komiyama A., Hashimoto T. Structural analysis of cDNAs for subunits of human mitochondrial fatty acid beta-oxidation trifunctional protein. Biochem Biophys Res Commun. 1994 Mar 15;199(2):818–825. doi: 10.1006/bbrc.1994.1302. [DOI] [PubMed] [Google Scholar]
- Kamijo T., Aoyama T., Miyazaki J., Hashimoto T. Molecular cloning of the cDNAs for the subunits of rat mitochondrial fatty acid beta-oxidation multienzyme complex. Structural and functional relationships to other mitochondrial and peroxisomal beta-oxidation enzymes. J Biol Chem. 1993 Dec 15;268(35):26452–26460. [PubMed] [Google Scholar]
- Kamijo T., Wanders R. J., Saudubray J. M., Aoyama T., Komiyama A., Hashimoto T. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J Clin Invest. 1994 Apr;93(4):1740–1747. doi: 10.1172/JCI117158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R. I., Morton D. H. 3-Hydroxyoctanoic aciduria: identification of a new organic acid in the urine of a patient with non-ketotic hypoglycemia. Clin Chim Acta. 1988 Jun 30;175(1):19–26. doi: 10.1016/0009-8981(88)90031-9. [DOI] [PubMed] [Google Scholar]
- Montell C., Berk A. J. Elimination of mRNA splicing by a point mutation outside the conserved GU at 5' splice sites. Nucleic Acids Res. 1984 May 11;12(9):3821–3827. doi: 10.1093/nar/12.9.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M., MacDonald C. C., Berget S. M. Are vertebrate exons scanned during splice-site selection? Nature. 1992 Nov 19;360(6401):277–280. doi: 10.1038/360277a0. [DOI] [PubMed] [Google Scholar]
- Orita M., Iwahana H., Kanazawa H., Hayashi K., Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci U S A. 1989 Apr;86(8):2766–2770. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N., Sherman J., Miller F., Mena H., Plotz P. A 5' splice junction mutation leading to exon deletion in an Ashkenazic Jewish family with phosphofructokinase deficiency (Tarui disease). J Biol Chem. 1993 Mar 5;268(7):4963–4967. [PubMed] [Google Scholar]
- Senapathy P., Shapiro M. B., Harris N. L. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- Sharp P. A. Split genes and RNA splicing. Cell. 1994 Jun 17;77(6):805–815. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Yokota I., Coates P. M., Strauss A. W., Kelly D. P., Zhang Z., Gregersen N., Andresen B. S., Matsubara Y., Curtis D. Mutations in the medium chain acyl-CoA dehydrogenase (MCAD) gene. Hum Mutat. 1992;1(4):271–279. doi: 10.1002/humu.1380010402. [DOI] [PubMed] [Google Scholar]
- Treem W. R., Rinaldo P., Hale D. E., Stanley C. A., Millington D. S., Hyams J. S., Jackson S., Turnbull D. M. Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology. 1994 Feb;19(2):339–345. [PubMed] [Google Scholar]
- Uchida Y., Izai K., Orii T., Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J Biol Chem. 1992 Jan 15;267(2):1034–1041. [PubMed] [Google Scholar]
- Venizelos N., Ijlst L., Wanders R. J., Hagenfeldt L. beta-Oxidation enzymes in fibroblasts from patients with 3-hydroxydicarboxylic aciduria. Pediatr Res. 1994 Jul;36(1 Pt 1):111–114. doi: 10.1203/00006450-199407001-00020. [DOI] [PubMed] [Google Scholar]
- Wanders R. J., Ijlst L., Duran M., Jakobs C., de Klerk J. B., Przyrembel H., Rocchiccioli F., Aubourg P. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: different clinical expression in three unrelated patients. J Inherit Metab Dis. 1991;14(3):325–328. doi: 10.1007/BF01811694. [DOI] [PubMed] [Google Scholar]
- Wilcken B., Carpenter K. H., Hammond J. Neonatal symptoms in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child. 1993 Sep;69(3 Spec No):292–294. doi: 10.1136/adc.69.3_spec_no.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken B., Leung K. C., Hammond J., Kamath R., Leonard J. V. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet. 1993 Feb 13;341(8842):407–408. doi: 10.1016/0140-6736(93)92993-4. [DOI] [PubMed] [Google Scholar]