

Abstract
Aims
Thrombopoietin (Tpo) is known for its ability to stimulate platelet production. However, it is currently unknown whether Tpo plays a physiological function in the heart.
Methods and results
We assessed the potential protective role of Tpo in vitro and in vivo in two rat models of myocardial ischaemia/reperfusion. Tpo receptor (c-mpl) message was detected in the heart using RT-PCR, and the Tpo receptor protein was detected using western blotting and immunohistochemistry. Tpo treatment immediately before ischaemia reduced myocardial necrosis, apoptosis, and decline in ventricular function following ischaemia/reperfusion in the rat in a concentration- and dose-dependent manner with an optimal concentration of 1.0 ng/mL in vitro and an optimal dose of 0.05 μg/kg iv in vivo. Tpo also reduced infarct size when given after the onset of ischaemia or at reperfusion. Tpo activated JAK-2 (Janus kinase-2) and p44 MAPK (mitogen-activated protein kinase) during reperfusion but not prior to ischaemia. Inhibition of JAK-2 (AG-490), p42/44 MAPK (PD98059), mitochondrial KATP channels (5-HD), and sarcolemmal KATP channels (HMR 1098) abolished Tpo-induced resistance to injury from myocardial ischaemia/reperfusion. AG-490, PD98059, 5-HD, and HMR1098 alone had no effect on cardioprotection. Treatment with a single dose of Tpo (0.05 or 1.0 μg/kg iv) did not result in the elevation of platelet count or haematocrit over a 16-day period.
Conclusion
A single treatment of Tpo confers cardioprotection through JAK-2, p42/44 MAPK, and KATP channels, suggesting a potential therapeutic role of Tpo in the treatment of injury resulting from myocardial ischaemia and reperfusion.
Keywords: Ischaemia, thrombopoietin, protein kinases, infarction, K-ATP channel
1. Introduction
Ischaemic heart disease is the underlying cause of most acute myocardial infarctions, congestive heart failure, arrhythmias, and sudden cardiac death.1 Protection of the heart against injury from ischaemia/reperfusion remains a challenge for the cardiologist and the cardiac surgeon. However, there are no current therapies that have been proved to directly protect the heart against the deleterious effects of ischaemia and reperfusion in humans. Recent studies have shown that erythropoietin (Epo), a cytokine used to stimulate red blood cell production, protects the heart against ischaemic injury2-4 by a mechanism that involves activation of pro-survival kinases and ATP-dependent potassium (KATP) channels.5 Thrombopoietin (Tpo), another cytokine, is the primary physiological regulator of megakaryocyte and platelet development exerting its action through the Tpo receptor, c-mpl. However, it is currently unknown whether Tpo plays a physiological function in the myocardium. Tpo shares ~25% similarity in sequence identity with Epo in the N-terminal domain. We hypothesized that Tpo would be able to protect the heart against injury caused by ischaemia/reperfusion by decreasing infarct size and apoptosis by enhancing the recovery of ventricular function after ischaemia. We selected in vitro and in vivo models of global and regional myocardial ischaemia and reperfusion to determine the efficacy of Tpo to confer cardioprotection in the rat. Furthermore, we determined the role of Janus kinases (JAKs), signal transducers and activators of transcription (STAT), p42/44 mitogen-activated protein kinase (MAPK), and KATP channels in mediating cardioprotection by Tpo and the impact of a single Tpo treatment on the long-term levels of platelets and haematocrit.
2. Methods
Male Sprague Dawley rats at 8 weeks of age used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Recombinant human Tpo was obtained from Leinco Technologies, St Louis, MO, USA.
2.1 Detection of thrombopoietin receptor (c-mpl) in the heart
Primers (forward primer: 5′-CTA GCT CCC GAG GCT TCT TC-3′; reverse primer: 5′-GGC TCC AGC ACC TTC CAG TCC-3′) for c-mpl were designed according to Rouleau et al.6 Adult rat ventricular cardiomyocytes were isolated from male Sprague Dawley rats as described previously.7 Heart homogenates were prepared and western analysis studies were performed using methods described previously.8 A polyclonal antibody specific to c-mpl was obtained from Santa Cruz Biotechnology, CA, USA, Cat No. sc-13189. Immunostaining was performed using the same anti-c-mpl antibody at 2 μg/mL and the Santa Cruz goat ABC Staining System. Prior to immunostaining, unfixed frozen tissue sections were thawed and allowed to air-dry at room temperature. Sections were then rehydrated for 15 min at room temperature in PBS containing 0.1% (w/v) Saponin (Sigma). Saponin was also added to the blocking solution, which was also used to dilute the primary antibody. Sections were incubated overnight at 4°C in primary antibody. All other steps were performed according to the kit protocol. Digital images were captured using a Nikon Eclipse E600 microscope equipped with Spot RT Slider Camera and software (Diagnostic Instruments, Inc.).
2.2 Thrombopoietin and cardioprotection studies in vitro
Isolated rat hearts were perfused retrogradely and instrumented as described previously.9 Hearts were subjected to 25 min of global no-flow ischaemia and 3 h of reperfusion.
2.3 Apoptosis measurements
Hearts from Sprague Dawley rats perfused continuously with bicarbonate buffer for 240 min served as non-ischaemic controls. Hearts aerobically perfused for 35 min and subjected to 25 min of global ischaemia followed by 3 h of reperfusion served as the ischaemia/reperfusion control group. Hearts aerobically perfused with buffer for 20 min and then perfused with Tpo (1 ng/mL) for 15 min before global ischaemia and reperfusion served as a Tpo-treated group. At the end of perfusion, each heart was snap-frozen in liquid nitrogen. Four frozen sections (10 μm thick) from the free wall of left ventricle of each heart (72 sections total) were processed for TUNEL assay according to the manufacturer’s protocol (Roche). The slides were examined under a fluorescent microscope (400× magnification, excitation 490 nm, and emission 515 nm). Twenty-two random high-power fields from each heart sample were chosen and blindly quantified. TUNEL results are presented as percentage (positive nuclei per field/total nuclei per field × 100).
2.4 Western immunoblotting studies
At 15 min after perfusion with Tpo and at 5 min after reperfusion, the free wall of the left ventricle of the heart was excised and snapfrozen in liquid nitrogen for JAK, STAT, and p42/44 MAPK analysis as described previously.10 Cell lysates were probed with specific antibodies against phosphorylated (Thr 202/Tyr 204) p42/44 MAPK, non-phosphorylated p42/44 MAPK (1:1000 dilution), phosphorylated JAK-2 (Tyr 1007/Tyr 1008), total JAK-2 (1:1000 dilution), phosphorylated STAT3 (Tyr 705) and STAT5 (Tyr 694) (1:1000 dilution), β-actin, or GAPDH (1:1000 dilution) overnight at 4°C and then incubated with horseradish peroxidase-conjugated secondary antibody in 5% non-fat milk in PBST for 1 h at room temperature. After the final washes, the membranes were incubated in enhanced chemiluminescence solution (Amersham Life Science, Arlington Heights, IL, USA) followed by exposure to chemiluminescence film.
2.5 Thrombopoietin and cardioprotection studies in vivo
An in vivo anesthetized rat model was used for these experiments, using the general surgical protocol and determination of infarct size described previously.11 For infarct size studies, rats underwent 30 min of regional ischaemia followed by 3 h of reperfusion. Different doses of Tpo were administered intravenously as a bolus 15 min before ischaemia.
2.6 Platelet and haematocrit levels following thrombopoietin treatment
Finally, rats were treated with a single dose of Tpo (0.05 or 1.0 μg/kg iv), and platelet count and haematocrit measured every 4 days over a 16-day follow-up period.
2.7 Statistical analysis
Data reported are mean ± standard error of the mean (SE). Statistical analysis was performed by the use of repeated measures ANOVA with the Greenhouse–Geisser adjustment used to correct for the inflated risk of a type I error.12 If significant, the Mann–Whitney or Dunnett’s test was used as a second step to identify which groups were significantly different.12 Significance was set at P < 0.05.
3. Results
3.1 Presence of the thrombopoietin receptor, c-mpl in the heart
Using primers based on the sequence for c-mpl, we amplified the message for the Tpo receptor in heart homogenates from rat, using RT-PCR. The sequence of the amplified PCR fragments for rat c-mpl was deposited in GenBank (accession number DQ013343). The sequence was homologous to the predicted sequence for rat present in GenBank (XM-001072502). The presence of c-mpl protein in heart homogenates and cardiomyocytes obtained from Sprague Dawley rats was then detected by western blotting. Human chronic myelogenous leukaemia cells were used as a positive control (Figure 1). To complement these studies, we performed immunohistochemistry to demonstrate the presence and distribution of c-mpl in the heart. Immunohistochemical staining revealed that c-mpl was distributed throughout the myocytes but not the vasculature (Figure 1).
Figure 1.
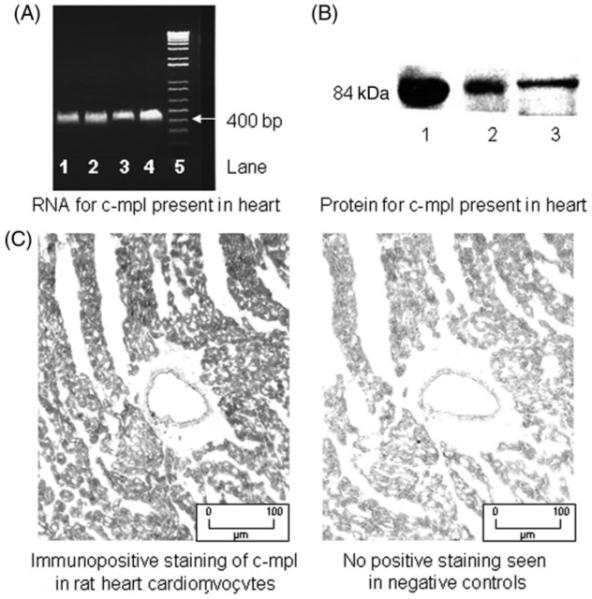
The presence of the thrombopoietin receptor c-mpl in the heart. (A) Determination of c-mpl mRNA by RT-PCR. Total mRNA was amplified by RT-PCR with specific primers, as described under Methods. c-mpl, lanes 1–4, was detected in four different rat hearts analysed. Lane 5 represents a 100 bp ladder standard. (B) Determination of c-mpl protein by immunoblot analysis of rat heart ventricles, using an anti c-mpl antibody. Lane 1 contains c-mpl from cardiomyocytes; lane 2, c-mpl from whole heart. Lane 3 is the positive control: human chronic myelogenous leukaemia cells. (C) Immunohistochemical localization of c-mpl in myocytes but not coronary vasculature. Data are representative of three analyses for each sample.
3.2 Thrombopoietin and cardioprotection studies in vitro
Rat hearts were isolated and perfused with Tpo at 0.01, 0.1, 1.0, or 10.0 ng/mL for 15 min prior to 25 min of global ischaemia and 180 min of reperfusion. Tpo (1.0 ng/mL) reduced coronary flow prior to ischaemia from 6 to 5 mL/min/g, increased left ventricular developed pressure (LVDP) from 119 ± 7 to 151 ± 8 mmHg, and decreased heart rate from 240 ± 17 to 228 ± 8 b.p.m. (Table 1). Tpo decreased post-ischaemic infarct size in a ‘U’-shaped concentration-dependent manner. The optimal concentration of Tpo that afforded a maximum reduction of infarct size was 1.0 ng/mL (Figure 2A). Tpo also increased the recovery of LVDP following ischaemia and reperfusion. Similar to the infarct size studies, the optimal concentration that afforded maximal recovery of post-ischaemic LVDP was 1.0 ng/mL (Figure 2A). Recovery of heart rate was decreased from 94 ± 3% in untreated hearts to 85 ± 4% of pre-ischaemic values in hearts treated with 1.0 ng/mL Tpo. Recovery of coronary flow was unaffected by Tpo. These data indicate that Tpo rapidly protects the isolated rat heart against injury from ischaemia–reperfusion as assessed by infarct size reduction and recovery of developed pressure in a concentration-dependent manner.
Table 1.
Haemodynamic values for thrombopoietin concentration–response studies in vitro
| Groups | Before drug administration |
After drug administration |
Reperfusion |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Heart rate (b.p.m.) |
Coronary flow rate (mL/min/ g) |
Left ventricular developed pressure (mmHg) |
Heart rate (b.p.m.) |
Coronary flow rate (mL/min/ g) |
Left ventricular developed pressure (mmHg) |
Heart rate (b.p.m.) |
Coronary flow rate (mL/min/ g) |
Left ventricular developed pressure (mmHg) |
|
| Drug-free control | 246 ± 10 | 6 ± 1 | 117 ± 6 | — | — | — | 232 ± 13 | 4 ± 1 | 39 ± 5 |
| Tpo (0.01 ng/mL) | 225 ± 16 | 6 ± 1 | 130 ± 7 | 202 ± 8 | 5 ± 1 | 156 ± 8 | 184 ± 16 | 3 ± 1 | 52 ± 6 |
| Tpo (0.1 ng/mL) | 228 ± 11 | 5 ± 1 | 120 ± 7 | 233 ± 7 | 5 ± 1 | 158 ± 8 | 197 ± 19 | 3 ± 1 | 64 ± 6* |
| Tpo (1.0 ng/mL) | 240 ± 17 | 6 ± 1 | 119 ± 7 | 228 ± 8 | 5 ± 1 | 151 ± 8 | 204 ± 22 | 4 ± 1 | 62 ± 5* |
| Tpo (10.0 ng/mL) | 226 ± 16 | 6 ± 1 | 134 ± 6 | 220 ± 7 | 6 ± 1 | 172 ± 7 | 183 ± 11 | 4 ± 1 | 64 ± 6* |
Tpo, thrombopoietin. Data are mean ± SE, n = 6 per group, *P < 0.05, Tpo vs. drug-free control.
Figure 2.

Thrombopoietin concentration–response studies in vitro. (A) Percent infarction and recovery of left ventricular developed pressure in the heart following 15 min of treatment with thrombopoietin at 0.01, 0.1, 1.0, or 10.0 ng/mL prior to 25 min of global ischaemia and 180 min of reperfusion. Data are means ± SE, n = 8 hearts per group. *P < 0.05, thrombopoietin vs. drug-free control. (B) Thrombopoietin limits apoptosis in vitro following a 15 min treatment (1.0 ng/mL) prior to 25 min of global ischaemia and 180 min of reperfusion. Quantification of TUNEL staining to demonstrate thrombopoietin decreases ischaemia/reperfusion-induced apoptosis. Data are means ± SE. *P < 0.05 vs. perfusion; #P < 0.05 vs. ischaemia/reperfusion.
3.3 Apoptosis studies
Since Tpo reduced infarct size and increased the recovery of LVDP, we also examined whether Tpo protects the ischaemic myocardium against apoptosis. Using TUNEL-labelled sections obtained from the left ventricle after 25 min of global ischaemia and 3 h of reperfusion, the extent of TUNEL-positive cells observed (3.56 ± 0.36%) was almost twice that observed in non-ischaemic hearts (1.91 ± 0.27%). In the Tpo-treated group, TUNEL staining was comparable (1.94 ± 0.26%, P < 0.05) with that observed in control hearts not subjected to ischaemia (Figure 2B). Representative TUNEL-stained sections are shown in Figure 2B demonstrating fewer apoptotic positive cells in the Tpo-treated heart.
3.4 Role of Janus kinase and signal transducers and activators of transcription in thrombopoietin-induced cardioprotection
Binding of Tpo to c-mpl in non-cardiac cell lines results in the activation of JAK-2 and STAT.13,14 However, it is unknown whether this mechanism is present in the heart. To determine further the biological effects of Tpo on cardioprotection at the molecular level, we examined the effect of Tpo on the activation of JAK-2. Tpo had no effect on phosphorylation of JAK-2 before ischaemia. Reperfusion induced phosphorylation of JAK-2 in untreated hearts; however, Tpo further increased phosphorylation of JAK-2 during reperfusion (Figure 3A). Tpo did not alter total JAK-2 levels before ischaemia or during reperfusion (Figure 3A). We then reasoned that if JAK-2 activation can be blocked with the JAK-2 inhibitor AG-490, Tpo-induced cardioprotection mediated through its receptor c-mpl would be prevented. Isolated rat hearts were perfused with AG-490 (1 μM) followed by Tpo prior to ischaemia/reperfusion. AG-490 alone had no effect on infarct size or recovery of developed pressure. However, treatment of hearts with AG-490 prior to perfusion in the presence of Tpo abolished its cardioprotective effect (Figure 3B). Inhibition of JAK-2 by AG-490 also abolished the cardioprotective effects of 0.1 and 10.0 ng/mL Tpo (results not shown). We then examined the effect of Tpo on the activation of STAT. STAT3 (Tyr 705) was activated by Tpo in the absence of ischaemia and was further activated by Tpo 5 min after reperfusion (Figure 3C). STAT5 (Tyr 694) was not activated by Tpo before ischaemia or during reperfusion (results not shown). These studies suggest Tpo acts through c-mpl, JAK-2, and STAT3 to confer protection against injury from ischaemia and reperfusion.
Figure 3.

Role of Janus kinase and signal transducers and activators of transcription in thrombopoietin-induced cardioprotection. (A) Western analysis of cell lysates from hearts demonstrates that thrombopoietin (1.0 ng/mL) does not cause phosphorylation of JAK-2 before ischaemia. Reperfusion induces phosphorylation of JAK-2. Thrombopoietin further increases phosphorylation of JAK-2 during reperfusion (upper panel). Bar graphs represent the mean ± SD. *P < 0.05 vs. perfusion. +P < 0.05, reperfusion plus thrombopoietin vs. reperfusion. (B) Percent infarction and recovery of left ventricular developed pressure following 15 min treatment with thrombopoietin (1.0 ng/mL) and a JAK-2 inhibitor prior to 25 min global ischaemia and 180 min reperfusion. The JAK-2 inhibitor was AG-490 (1 μM). Data are means ± SE, n = 6 hearts per group. †P < 0.05, thrombopoietin vs. drug-free control; #P < 0.05, thrombopoietin plus AG-490 vs. thrombopoietin. (C) Western analysis of cell lystates demonstrates that thrombopoietin phosphorylates STAT3 prior to ischaemia and further increases phosphorylation of STAT3 during reperfusion. Bar graphs represent the mean ± SD. *P < 0.05, vs. perfusion; +P < 0.95, reperfusion plus thrombopoietin vs. reperfusion.
3.5 Role of p42/44 MAPK in thrombopoietin-induced cardioprotection
The pro-survival kinase p42/44 MAPK is an important mediator of cardioprotection.15,16 To determine whether Tpo-induced cardioprotection is mediated by p42/44 MAPK, hearts were perfused with the p42/44 MAPK inhibitor PD98059 alone for 15 min or in combination with Tpo (1.0 ng/mL) for another 15 min prior to ischaemia and reperfusion. PD98059 (10 μM) abrogated the effect of Tpo to reduce myocardial necrosis and to enhance ventricular function following ischaemia/reperfusion. PD98059 alone had no effect on cardioprotection (Figure 4A). Inhibition of p42/44 MAPK by PD98059 also abolished the cardioprotective effects of 0.1 and 10.0 ng/mL Tpo (results not shown). To corroborate the involvement of p42/44 MAPK in Tpo-induced cardioprotection, western analysis studies were then performed. Tpo had no effect on phosphorylation of p42/44 MAPK before ischaemia. However, although reperfusion itself induced phosphorylation of p42 MAPK and p44 MAPK, Tpo further increased the extent of phosphorylation of p44 MAPK but not p42 MAPK during reperfusion (Figure 4B). Our data suggest that the cardioprotective effects of Tpo are at least partially mediated by p44 MAPK.
Figure 4.

p42/44 MAPK-mediated cardioprotective effects of thrombopoietin. (A) Percent infarction and recovery of left ventricular developed pressure following a 15 min treatment with thrombopoietin (1.0 ng/mL) and a p42/44 MAPK inhibitor prior to 25 min of global ischaemia and 180 min of reperfusion. The p42/44 MAPK inhibitor was PD98059 (10 μM). Data are means ± SE (n = 8 hearts per group). *P < 0.05, thrombopoietin vs. drug-free control. +P < 0.05, thrombopoietin plus PD98059 vs. thrombopoietin. (B) Western analysis of cell lysates from hearts demonstrates that thrombopoietin (1.0 ng/mL) does not cause phosphorylation of p42/44 MAPK prior to ischaemia. Reperfusion induces phosphorylation of p42 MAPK and p44 MAPK. Thrombopoietin further increases phosphorylation of p44 MAPK but not p42 MAPK during reperfusion (upper panel). Results with antibodies to non-phosphorylated p42/44 MAPK shown in the lower panel. Bar graphs represent the mean ± SE from three hearts. †P < 0.05, vs. perfusion. #P < 0.05, reperfusion plus thrombopoietin vs. reperfusion.
3.6 Thrombopoietin and cardioprotection studies in vivo
To assess the effects of Tpo in vivo, we used a rat model of regional ischaemia/reperfusion. Heart rate, mean arterial pressure, and the rate pressure product were quantified during baseline, 15 min into ischaemia, and at 3 h of reperfusion and compared with rats subjected to ischaemia without Tpo (Table 2). Significant differences in depression of mean arterial pressure were found for some groups when compared with drug-free hearts subjected to ischaemia and reperfusion. For each group, the area at risk compared with total left ventricle weight was calculated. There were no significant differences seen between groups (data not shown). Tpo decreased post-ischaemic infarct size in a ‘U’-shaped dose-dependent manner. The dose of Tpo that resulted in the maximum reduction of infarct size was 0.05 μg/kg (Figure 5A).
Table 2.
Haemodynamic values for thrombopoietin acute dose–response studies in vivo
| n | Baseline |
15 min Ischaemia |
3 h Reperfusion |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Heart rate (b.p.m.) |
MAP (mmHg) |
RPP (mmHg/s) |
Heart rate (b.p.m.) |
MAP (mmHg) |
RPP (mmHg/s) |
Heart rate (b.p.m.) |
MAP (mmHg) |
RPP (mmHg/s) |
||
| Ischaemia alone | 6 | 352 ± 7 | 136 ± 5 | 48 ± 2 | 361 ± 8 | 102 ± 4 | 36 ± 5 | 317 ± 7 | 89 ± 4 | 30 ± 6 |
| Tpo (0.005 μg/kg) | 6 | 386 ± 8 | 133 ± 7 | 50 ± 6 | 374 ± 7 | 104 ± 6 | 38 ± 9 | 359 ± 6 | 92 ± 5 | 32 ± 5 |
| Tpo (0.05 μg/kg) | 6 | 342 ± 7 | 138 ± 5 | 50 ± 3 | 352 ± 6 | 124 ± 6 | 47 ± 5 | 332 ± 7 | 105 ± 5* | 38 ± 4 |
| Tpo (0.5 μg/kg) | 6 | 371 ± 6 | 144 ± 5 | 49 ± 3 | 382 ± 7 | 121 ± 7 | 44 ± 6 | 364 ± 7 | 108 ± 7* | 37 ± 5 |
Tpo, thrombopoietin. Data are mean ± SE, n = 6 per group, * P < 0.05, thrombopoietin plus ischaemia vs. ischaemia alone. MAP, mean arterial pressure; RPP, rate pressure product.
Figure 5.
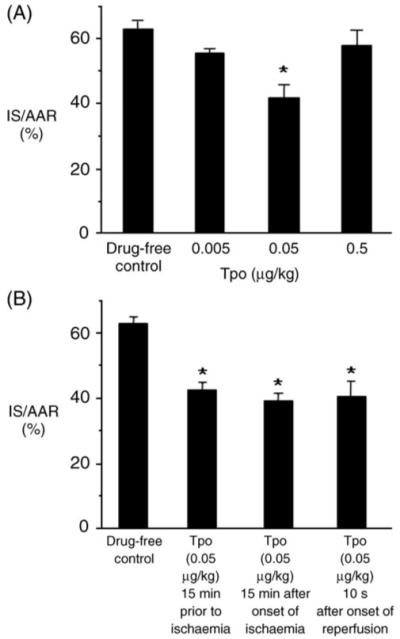
(A) Thrombopoietin dose–response studies in vivo. Percent infarction in the left ventricle following a 15 min treatment with thrombopoietin (0.005, 0.05, or 0.5 μg/kg iv) prior to 30 min of regional ischaemia and 180 min of reperfusion. (B) Phase of action of thrombopoietin. Infarct size was determined after 30 min regional ischaemia and 120 min reperfusion. Data are mean ± SD, n = 6 hearts per group. *P < 0.05, thrombopoietin vs. drug-free control.
3.7 Timing of thrombopoietin administration
Clinically, patients generally receive medical treatment for acute myocardial infarction after the onset of symptoms. Thus, we determined whether Tpo is able to protect the heart against injury after the onset of ischaemia or when administered at the time of reperfusion. Rats were treated with a bolus of Tpo (0.05 μg/kg iv) 15 min prior to ischaemia, 15 min after the onset of ischaemia, or 10 s after the onset of reperfusion. Tpo was able to reduce infarct size when administered before and during ischaemia or immediately at the onset of reperfusion (Figure 5B).
3.8 Role of KATP channels in thrombopoietin-induced cardioprotection
KATP channels, highly expressed in myocardial sarcolemma and thought to be expressed in myocardial mitochondria, have been found to serve as triggers and mediators of cardioprotection.17 To investigate a role for KATP channels in mediating Tpo-induced cardioprotection, rats were treated with and without Tpo (0.05 μg/kg) 15 min prior to 30 min regional myocardial ischaemia and 120 min reperfusion. The mitochondrial KATP channel blocker 5-HD (10 mg/kg) or sarcolemmal KATP channel blocker HMR1098 (3 mg/kg) was administered either 20 min prior to ischaemia or 5 min prior to reperfusion. Prior administration of 5-HD or HMR1098 before ischaemia abolished infarct size reduction with Tpo. 5-HD and HMR1098 given alone before ischaemia had no effect on infarct size (Figure 6A). Administration of 5-HD but not HMR1098 prior to reperfusion abolished infarct size reduction produced by Tpo (Figure 6B). Administration of 5-HD and HMR1098 alone prior to reperfusion had no effect on infarct size. We then determined whether inhibition of the sarcolemmal KATP channel prevented Tpo-induced activation of JAK-2 and p42/44 MAPK. Rats were treated with the sarcolemmal KATP channel blocker HMR 1098 prior to administration of Tpo. Rats treated with Tpo alone, HMR 1098 alone, and no drug served as controls. The free wall of the left ventricle was harvested 5 min after reperfusion for western analysis of phosphorylated JAK-2 and p42/44 MAPK. Inhibition of the sarcolemmal KATP channel prevented Tpo-induced activation of JAK-2 and p44 MAPK (Figure 6C).
Figure 6.
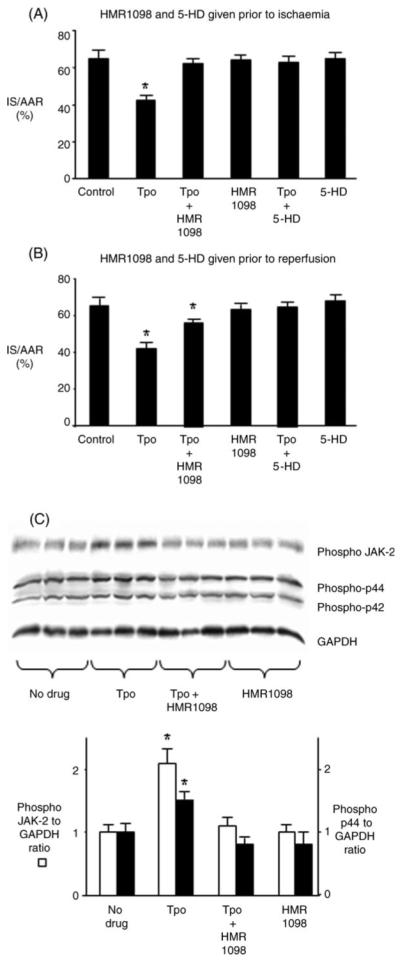
Potassium channel-mediated cardioprotective effects of thrombopoietin. Rats were treated with and without thrombopoietin (0.05 μg/kg) 15 min prior to 30 min of regional ischaemia and 180 min of reperfusion. HMR1098 (3 mg/kg) or 5-HD (10 mg/kg) was administered either 20 min prior to ischaemia (A) or 5 min prior to reperfusion (B). Data are mean ± SD, n = 6 per group. *P < 0.05, vs. control. (C) Western analysis of cell lysates demonstrates that HMR 1098 prevents phosphorylation of JAK-2 and p44/MAPK by thrombopoietin. Data are mean ± SD, n = 3 per group. *P < 0.05, vs. no drug.
3.9 Platelet and haematocrit levels following thrombopoietin treatment
To determine whether a single dose of Tpo that confers immediate cardioprotection in vivo resulted in an increased platelet count, rats were treated with Tpo (0.05 or 1.0 μg/kg iv). Prior to treatment, platelet counts in untreated rats were 850 ± 96 × 103/μL, and the haematocrit in untreated rats was 43.8 ± 1.8%. Following Tpo treatment, these values remained unchanged over the 16-day follow-up period (Table 3).
Table 3.
Platelet count and haematocrit following a single intravenous treatment with thrombopoietin
| Day |
|||||
|---|---|---|---|---|---|
| 0 | 4 | 8 | 12 | 16 | |
| Tpo (0.05 μg/kg) | |||||
| Platelet count (×103/μL) | 850 ± 96 | 820 ± 98 | 896 ± 111 | 805 ± 88 | 890 ± 150 |
| Haematocrit (%) | 43.8 ± 1.8 | 40.7 ± 0.9 | 43.6 ± 1.1 | 43.8 ± 2.3 | 42.6 ± 1.1 |
| Tpo (1.0 μg/kg) | |||||
| Platelet count (×103/μL) | 862 ± 98 | 851 ± 92 | 867 ± 101 | 852 ± 81 | 872 ± 102 |
| Haematocrit (%) | 43.2 ± 1.9 | 42.1 ± 1.2 | 43.6 ± 1.3 | 43.6 ± 1.9 | 43.8 ± 2.1 |
Data are mean ± SD, n = 5–6 per group.
4. Discussion
Our study demonstrates that the receptor for Tpo (c-mpl) is present in the heart and localized to the myocytes. We found that a single treatment with Tpo before ischaemia exerts an immediate protective effect against injury from myocardial ischaemia/reperfusion both in vitro and in vivo as manifested by a reduction in necrosis and apoptosis and an increase in the recovery of ventricular function following ischaemia. This immediate cardioprotective effect of Tpo is concentration- and dose-dependent with optimal protection occurring at 1.0 ng/mL in vitro and 0.05 μg/kg iv in vivo. Tpo also reduced infarct size when given after the onset of ischaemia or at reperfusion. Increased resistance to injury from myocardial ischaemia/reperfusion conferred by Tpo is mediated by JAK-2, p42/44 MAPK, and KATP channels. Furthermore, increased resistance to injury is observed immediately after treatment with Tpo, indicating that the induction of new genes is not necessary for its cardioprotective effect to be manifested. Tpo confers its protective effect against injury from myocardial ischaemia/reperfusion without increasing platelet levels or haematocrit. Our finding that Tpo is cardioprotective in the isolated buffer-perfused heart suggests that it has direct cardioprotective properties, independent of its ability to promote platelet production. These original observations may cause a paradigm shift in how we view the mechanisms and biological effects of Tpo. In support of this idea, Tpo has been shown to act as a protective agent against doxorubicin-induced cardiac injury.18
In platelets, activation of c-mpl by Tpo induces secondary tyrosine kinase activity in cytoplasmic proteins and recruits cytoplasmic JAK-2 resulting in phosphorylation of specific tyrosine residues on c-mpl.13 We show Tpo acts via activation of JAK-2 in hearts, suggesting signalling through c-mpl is necessary to confer cardioprotection. Tpo also activates STAT3 within minutes following administration, suggesting the signalling pathway stimulated by Tpo is capable of conducting a signal to the nucleus. Further studies are needed to determine the events following Tpo-induced STAT activation and translocation to the nucleus and their role in cardioprotection.
The signalling pathway by which Tpo protects against injury to the heart caused by ischaemia is mediated in part by p42/44 MAPK and KATP channels. Tpo increased phosphorylation of p44 but not p42 MAPK during reperfusion with cardioprotection produced by Tpo abrogated by the p42/44 MAPK inhibitor PD98059. p42/44 MAPK activation is known to reduce ischaemia/reperfusion injury in the heart and to upregulate the bcl family of anti-apoptotic genes. In support of this, we demonstrated that Tpo reduced the extent of apoptosis induced by ischaemia and reperfusion by the TUNEL assay. Activation of KATP channels is also associated with increased resistance to injury from myocardial ischaemia/reperfusion conferred by ischaemic and pharmacological pre-conditioning. Currently there are two KATP channels thought to be present in the cardiac myocyte, one in the sarcolemma (sarc KATP channel) and one in the inner mitochondrial membrane (mito KATP channel). Our data show that the sarcolemmal KATP channel appears to act as trigger and the mitochondrial KATP channel as an effector of Tpo-induced cardioprotection. There is evidence to suggest that opening either channel may be important in producing cardioprotection, although the bulk of evidence suggests that it is the mito KATP channel that is responsible for mediating the protective effect of ischaemic pre-conditioning.19 Inhibition of the sarc KATP channel prevented activation of JAK-2 and p44 MAPK, supporting the notion that the sarc KATP channel acts as a trigger for Tpo-induced cardioprotection. The role of additional components in the signalling pathway by which the heart is protected by Tpo against injury remains unknown.
Tpo also confers protection against injury when given after the onset of ischaemia, i.e. after the onset of symptoms. In patients experiencing symptoms of a myocardial infarction, or those who are about to undergo cardiac surgery, Tpo could be administered acutely to decrease injury to the heart from ischaemia/reperfusion. Thus, Tpo may represent an important and potent agent for immediately increasing cardioprotection. Cardioprotection by ischaemic pre-conditioning is a powerful endogenous phenomenon in which brief episodes of a subtoxic ischaemic insult induce robust protection against more prolonged, lethal ischaemia. The molecular mechanisms underlying ischaemic pre-conditioning are still being elucidated and clinical application remains elusive, as it has not yet gained widespread acceptance as a treatment strategy. Pharmacological pre-conditioning against ischaemia could offer a more practical way of harnessing the molecular mechanisms responsible for increased cardioprotection. Our study shows that pharmacological pre-conditioning through Tpo is effective and represents a novel cardioprotective strategy in the setting of elective myocardial ischaemia and reperfusion as encountered during cardiac surgery and during acute myocardial infarction. Tpo has been evaluated in patients with cancer who require platelet transfusion support. Furthermore, SB-497115, an oral Tpo receptor agonist, is currently undergoing phase II and III clinical trials in adults receiving chemotherapy for advanced solid tumours and in immune thrombocytopenic purpura. AMG531, an analogue of Tpo, is currently being evaluated in a phase III study for the treatment of thrombocytopenia.
The dose of Tpo used in vivo of 0.05 μg/kg to confer immediate cardioprotection is approximately 10 times lower than that used in humans to stimulate platelet production in cancer patients20 and to mobilize peripheral blood progenitor cells.21,22 In initial human trials of Tpo, doses of 1–5 μg/kg increased platelet production five to 10 times in healthy individuals and in those about to receive chemotherapy for malignancy. Similarly in rats, the dose of PEGylated Tpo used to reduce thrombocytopenia (100 μg/kg)23 and to ameliorate thrombocytopenia in carboplatin-treated rats (1–30 μg/kg)24 is also considerably higher than the dose used in our study. The single dose of Tpo (0.05 or 1.0 μg/kg) we used did not result in an increased platelet count or haematocrit over the 16-day follow-up period.
In summary, a single treatment with Tpo confers immediate protection against injury caused by ischaemia–reperfusion in the heart, with protection mediated by JAK-2, STAT3, p42/44 MAPK, and KATP channels. The level of protection conferred by Tpo is comparable with that achieved by ischaemic pre-conditioning and is manifest at a dose that does not result in increased platelet count or haematocrit. Further studies are warranted to define the pathways by which binding of Tpo to c-mpl confers cardioprotection. Our results suggest that Tpo directly protects the heart and may represent a novel approach for the treatment of acute myocardial infarction.
Acknowledgements
The technical help offered by Eugene Konorev and Jingli Wang and the secretarial assistance of Mary Lynne Koenig are gratefully acknowledged.
Funding
This study was supported in part by grant HL 54075 to J.E.B. and HL 08311 to G.J.G.
Footnotes
Conflict of interest: none declared.
References
- 1.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 2.Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, et al. Recombinant human erythropoietin protects the myocardium from ischaemia–reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci USA. 2003;100:4802–4806. doi: 10.1073/pnas.0630444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, et al. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci USA. 2003;100:11612–11617. doi: 10.1073/pnas.1930406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, et al. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999–1007. doi: 10.1172/JCI18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi Y, Rafiee P, Su J, Pritchard K, Jr, Tweddell J, Baker J. Acute cardioprotective effects of erythropoietin in infant rabbits are mediated by activation of protein kinases and potassium channels. Basic Res Cardiol. 2004;99:173–182. doi: 10.1007/s00395-004-0455-x. [DOI] [PubMed] [Google Scholar]
- 6.Rouleau C, Cui K, Feldman L. A functional erythropoietin receptor is necessary for the action of thrombopoietin on erythroid cells lacking c-mpl. Exp Hematol. 2004;32:140–148. doi: 10.1016/j.exphem.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Konorev EA, Kennedy MC, Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch Biochem Biophys. 1999;368:421–428. doi: 10.1006/abbi.1999.1337. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y, Baker JE, Zhang C, Tweddell JS, Su J, Pritchard KA., Jr. Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circ Res. 2002;91:300–306. doi: 10.1161/01.res.0000031799.12850.1e. [DOI] [PubMed] [Google Scholar]
- 9.Baker JE, Konorev EA, Gross GJ, Chilian WM, Jacob HJ. Resistance to myocardial ischemia in five rat strains: is there a genetic component of cardioprotection? Am J Physiol Heart Circ Physiol. 2000;278:H1395–H1400. doi: 10.1152/ajpheart.2000.278.4.H1395. [DOI] [PubMed] [Google Scholar]
- 10.Baker J, Hsu A, Fu X, Kozik D, Tweddell J, Gross G. Darbepoetin alfa protects the rat heart against infarction: dose–response, phase of action and mechanisms. J Cardiovasc Pharmacol. 2007;49:337–345. doi: 10.1097/FJC.0b013e318040cf81. [DOI] [PubMed] [Google Scholar]
- 11.Patel HH, Fryer RM, Gross ER, Bundey RA, Hsu AK, Isbell M, et al. 12-lipoxygenase in opioid-induced delayed cardioprotection: gene array, mass spectrometric, and pharmacological analyses. Circ Res. 2003;92:676–682. doi: 10.1161/01.RES.0000065167.52922.F6. [DOI] [PubMed] [Google Scholar]
- 12.Baker JE, Holman P, Gross GJ. Preconditioning in immature rabbit hearts: role of KATP channels. Circulation. 1999;99:1249–1254. doi: 10.1161/01.cir.99.9.1249. [DOI] [PubMed] [Google Scholar]
- 13.Drachman JG, Millett KM, Kaushansky K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J Biol Chem. 1999;274:13480–13484. doi: 10.1074/jbc.274.19.13480. [DOI] [PubMed] [Google Scholar]
- 14.Gurney AL, Wong SC, Henzel WJ, de Sauvage FJ. Distinct regions of c-Mpl cytoplasmic domain are coupled to the JAK–STAT signal transduction pathway and Shc phosphorylation. Proc Natl Acad Sci USA. 1995;92:5292–5296. doi: 10.1073/pnas.92.12.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafiee P, Shi Y, Kong X, Pritchard K, Jr, Tweddell J, Litwin S, et al. Activation of protein kinases in chronically hypoxic infant human and rabbit hearts: role in cardioprotection. Circulation. 2002;106:239–245. doi: 10.1161/01.cir.0000022018.68965.6d. [DOI] [PubMed] [Google Scholar]
- 16.Rafiee P, Shi Y, Su J, Pritchard K, Jr, Tweddell JS, Baker JE. Erythropoietin protects the infant heart against ischemia–reperfusion injury by triggering multiple signaling pathways. Basic Res Cardiol. 2005;100:187–197. doi: 10.1007/s00395-004-0508-1. [DOI] [PubMed] [Google Scholar]
- 17.Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol. 2003;285:H921–H930. doi: 10.1152/ajpheart.00421.2003. [DOI] [PubMed] [Google Scholar]
- 18.Li K, Sung RY, Huang WZ, Yang M, Pong NH, Lee SM, et al. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006;113:2211–2220. doi: 10.1161/CIRCULATIONAHA.105.560250. [DOI] [PubMed] [Google Scholar]
- 19.Hanley PJ, Daut J. K(ATP) channels and preconditioning: a re-examination of the role of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Vadhan-Raj S, Murray LJ, Bueso-Ramos C, Patel S, Reddy SP, Hoots WK, et al. Stimulation of megakaryocyte and platelet production by a single dose of recombinant human thrombopoietin in patients with cancer. Ann Intern Med. 1997;126:673–681. doi: 10.7326/0003-4819-126-9-199705010-00001. [DOI] [PubMed] [Google Scholar]
- 21.Gajewski JL, Rondon G, Donato ML, Anderlini P, Korbling M, Ippoliti C, et al. Use of thrombopoietin in combination with chemotherapy and granulocyte colony-stimulating factor for peripheral blood progenitor cell mobilization. Biol Blood Marrow Transplant. 2002;8:550–556. doi: 10.1053/bbmt.2002.v8.pm12434950. [DOI] [PubMed] [Google Scholar]
- 22.Linker C, Anderlini P, Herzig R, Christiansen N, Somlo G, Bensinger W, et al. Recombinant human thrombopoietin augments mobilization of peripheral blood progenitor cells for autologous transplantation. Biol Blood Marrow Transplant. 2003;9:405–413. doi: 10.1016/s1083-8791(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 23.Harada K, Tazunoki Y, Ide Y, Takeuchi A, Kawahara J, Suzuki T. Effects of pegylated recombinant human megakaryocyte growth and development factor on 5-fluorouracil-induced thrombocytopenia in balloon-injured rats. J Pharm Pharmacol. 2000;52:321–325. doi: 10.1211/0022357001773869. [DOI] [PubMed] [Google Scholar]
- 24.Ide Y, Harada K, Imai A, Yanagida M. PEG-rHuMGDF ameliorates thrombocytopenia in carboplatin-treated rats without inducing myelofibrosis. Int J Hematol. 1999;70:91–96. [PubMed] [Google Scholar]