

Abstract
Heart valve malformations are one of the most common types of birth defects, illustrating the complex nature of valve development. Vascular endothelial growth factor (VEGF) signaling is one pathway implicated in valve formation, however its specific spatial and temporal roles remain poorly defined. To decipher these contributions, we use two inducible dominant negative approaches in mice to disrupt VEGF signaling at different stages of embryogenesis. At an early step in valve development, VEGF signals are required for the full transformation of endocardial cells to mesenchymal cells (EMT) at the outflow tract (OFT) but not atrioventricular canal (AVC) endocardial cushions. This role likely involves signaling mediated by VEGF receptor 1 (VEGFR1), which is highly expressed in early cushion endocardium before becoming downregulated after EMT. In contrast, VEGFR2 does not exhibit robust cushion endocardium expression until after EMT is complete. At this point, VEGF signaling acts through VEGFR2 to direct the morphogenesis of the AVC cushions into mature, elongated valve leaflets. This latter role of VEGF requires the VEGF-modulating microRNA, miR-126. Thus, VEGF roles in the developing valves are dynamic, transitioning from a differentiation role directed by VEGFR1 in the OFT to a morphogenetic role through VEGFR2 primarily in the AVC-derived valves.
Keywords: VEGF, Heart valve development, Endocardial-mesenchymal transformation, endocardial cushions, outflow tract, atrioventricular canal, mitral valve, NFATc1, miR-126
Introduction
Congenital heart valve defects occur in a remarkable 2-3% of the human population (Brickner et al., 2000; Pierpont et al., 2007). While many abnormalities do not become apparent until adulthood, their origins lie in aberrant molecular and cellular events during embryogenesis. Heart valve development is a multi-step process initiated when endocardial cells within the endocardial cushions delaminate from the endocardium and transdifferentiate into mesenchymal cells. This endocardial-to-mesenchymal (EMT) process occurs in both the outflow tract (OFT) and atrioventricular canal (AVC) cushions at embryonic day 9.5 (E9.5) during mouse development. EMT initiates with the exchange of signals between the myocardium and endocardium (Runyan and Markwald, 1983) across a specialized expanse of extracellular matrix (the cardiac jelly) (Manasek et al., 1973) produced by myocardial cells (Krug et al., 1985). Once induced to undergo EMT, endocardial cells delaminate from the epithelium and transform into mesenchymal cells that invade and migrate through the cardiac jelly (Markwald et al., 1977). EMT must be subsequently terminated and proliferation of cushion mesenchymal cells attenuated to prevent valve hyperplasia. After EMT, a second process occurs in which the endocardial cushion area elongates and undergoes continuous remodeling that eventually refines the primitive cushions into thin, elongated valve leaflets. Each of these steps of valve development requires coordinated but poorly understood signaling between myocardial, endocardial, and cushion mesenchymal cells.
Vascular endothelial growth factor (VEGF) signaling, while best known for its pro-angiogenic role as mediated by VEGFA, is one pathway implicated in mammalian valve formation (Chang et al., 2004; Dor et al., 2001). VEGF signaling is activated by a combination of five ligands (VEGFA, VEGFB, placental growth factor (PlGF), VEGFC, and VEGFD) that bind to two primary receptors (VEGFR1 (Flt) and VEGFR2 (KDR, Flk)) with different specificities (reviewed in (Ferrara et al., 2003)). Further, Neuropilin (Nrp) proteins act as VEGF co-receptors (Soker et al., 1998) and an array of molecules, including the microRNA miR-126 (Fish et al., 2008; Kuhnert et al., 2008; Liu et al., 2009), modulate the VEGF pathway both extra- and intracellularly. The various VEGF ligands can produce different responses or have overlapping functions while the receptors can produce opposing effects or act in concert depending on the cellular context. Therefore, there can be a tremendous diversity of cellular responses to VEGF signaling.
Genetic disruption of various components of VEGF signaling in mice produce an array of developmental cardiac abnormalities, consistent with the known expression of multiple VEGF family members in the developing heart. For example, deletion of VEGFR1 (Fong et al., 1995), VEGFR2 (Shalaby et al., 1995), or even one allele of VEGFA results in cardiovascular abnormalities and embryonic lethality (Carmeliet et al., 1996; Ferrara et al., 1996). Myocardial-specific deletion of VEGFA results in both heart muscle and endocardial defects (Giordano et al., 2001; Haigh et al., 2000). Transgenic mice with a 2-3 fold increase in VEGFA levels develop an abnormal myocardium and incompletely septated heart chambers (Miquerol et al., 2000). VEGF has also been implicated in DiGeorge syndrome, as removal of the VEGFA-164 isoform in mice causes phenotypes reminiscent of this congenital disease, including aberrant aortic arch artery patterning and outflow tract anomalies (Stalmans et al., 2003).
The molecular effectors of VEGF signaling that fulfill its requirements in heart development may include the calcineurin/nuclear factor of activated T-cells (NFAT) pathway. Lethal defects in heart valve formation occur in mice lacking the endocardial-expressed NFATc1 gene (de la Pompa et al., 1998; Ranger et al., 1998). At E11.5, NFATc1, as activated by its dephosphorylation and nuclear import induced by the Ca2+-sensitive phosphatase, calcineurin (Crabtree and Olson, 2002), directs remodeling of immature endocardial cushions into valve leaflets (Chang et al., 2004). Both NFAT and VEGF signaling also serve an earlier role in valve development, during the EMT process. At E9.5, calcineurin-NFATc2/c3/c4 signaling in the myocardium represses expression of VEGFA, a potent inhibitor of EMT (Chang et al., 2004). The normal increase in cushion myocardial VEGFA levels following EMT and the suppression of EMT by transgenic overexpression of VEGFA have led to the notion that it acts to prevent excessive EMT and valve hyperplasia (Chang et al., 2004; Dor et al., 2001). Because VEGF activates NFATc signaling in human postnatal endothelial cells (Armesilla et al., 1999; Hernandez et al., 2001; Johnson et al., 2003), VEGF is thought to likewise regulate the endocardial NFATc1 activity that is essential for valve remodeling. These proposed requirements for VEGF signaling in valve development have not been fully addressed in vivo, partially due to early lethality upon deletion of VEGFA, VEGFR1, and VEGFR2 and potential genetic redundancy between VEGF ligands.
To examine the in vivo developmental roles of VEGF signaling, we apply two inducible dominant negative approaches in transgenic mice. These genetic methods are designed to provide temporally controlled inhibition of different subsets of VEGF family members, overcoming potential redundancy and allowing the assignment of functions to subsets of VEGF ligands. We show that VEGF has distinct roles in regulating terminal differentiation of endocardial cells during EMT at the OFT and, later, the morphogenesis of maturing valve leaflets. Expression analysis suggests cushion endocardial VEGFR1 contributes to the differentiation role and VEGFR2 to the morphogenetic one. Unexpectedly, VEGF signaling requirements do not interface directly with the nuclear translocation of NFATc1. Further, we demonstrate that a VEGF-modulating microRNA, miR-126, is required for valve elongation, expanding the developmental processes this class of molecules controls during mammalian development. Thus, VEGF signaling in valve development involves distinct sets of ligands, receptors, and intracellular modulators, whose dynamic expression contributes to changing cellular roles and VEGF responses at different stages of heart valve development.
Materials and Methods
Establishment of TRE-sFlt Transgenic Mice
A sFlt-HA cassette (Jacobi et al., 2004) was subcloned into the pTRE2 vector (Clontech, Mountain View, CA). Pronuclear injection of the linearized plasmid with prokaryotic sequence removed was used to generate transgenic mouse lines. Mouse embryonic fibroblasts (MEFs) were produced for each line using a protocol described in (Stankunas et al., 2003). An actin-rtTA expressing plasmid, pCAGS-rtTA (Sarin et al., 2005), was transfected into the MEFs using FuGENE 6 (Roche Applied Science, Indianapolis, IN). One of the lines that produced a doxycycline (dox)-dependent increase in secreted sFlt-HA upon co-expression of rtTA was selected for all in vivo experiments.
Doxycycline Treatments
Dox treatment of pregnant mice were performed by a combination of intraperitoneal (IP) injections at 40 mg/kg and 100 μg/mL provided in the drinking water. IP injections were repeated once per day until embryos were harvested. Therefore, embryos were continuously exposed to dox during indicated periods. Embryo dates were assigned by monitoring vaginal plugs and ultrasonography (Chang et al., 2003), with corrections made based on the developmental morphology of non-experimental embryos at the time of harvesting.
Histology
Immediately after harvesting, embryos were fixed overnight at 4°C in 4% paraformaldehyde in phosphate-buffered saline (PBS). Embryos were then dehydrated through a sequential ethanol series, cleared with xylenes, and immersed overnight in melted paraffin wax. Paraffinized embryos were embedded in transverse orientation, sectioned to 7 μm slices using a Leica microtome, and transferred to microscope slides. Slides were rehydrated, stained sequentially with hematoxylin and eosin (H&E) (Polysciences, Warrington, PA), dehydrated, and mounted using Permount (Fisher, Pittsburgh, PA). Stained slides were imaged using a Nikon 90i widefield microscope and cardiac structures were analyzed as described previously (Stankunas et al., 2008b).
Immunostaining
Paraffin sections were prepared as described above and immunostaining was performed following published protocols (Chang et al., 2008). After rehydration, different antigen retrieval methods were used depending on the primary antibody. For anti-VEGFR2 (R&D Systems, Minneapolis, MN), sections were incubated at room temperature in 20 μg/mL Proteinase K for 10 minutes. For anti-PECAM (BD Biosciences, San Jose, CA), anti-smooth muscle actin (Sigma, St. Louis, MO), and anti-phospho-histone H3 (Upstate, Billerica, MA), sections were exposed to 0.05% trypsin (Invitrogen, Carlsbad, CA) for 45 minutes at 37°C. For anti-FoxP1 (Cell Signaling, Danvers, MA), sections were boiled for 10 minutes in a 0.5 mM EDTA, 10 mM Tris pH 9.5 solution. For anti-Sox9 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-NFATc1 (Santa Cruz Biotechnology), sections were boiled for 10 minutes in 0.3M citrate, pH 6.0 buffer (Vector Laboratories, Burlingame, CA). Following antigen retrieval, sections were blocked with either 5% normal goat serum in PBS or with M.O.M. blocking reagents (Vector Laboratories). Primary antibodies were diluted in blocking solution and left on sections overnight at 4°C. For immunofluorescence, staining was visualized using Alexa-dye conjugated secondary antibodies (Invitrogen) or reagents from the M.O.M. kit (Vector Laboratories). Nuclei were counterstained with Hoechst and slides mounted using Hardset Mounting Medium (Vector Laboratories). For immunohistochemistry, staining was visualized using biotinylated secondary antibodies (Jackson Immunoresearch, West Grove, PA), streptavidin-conjugated horse radish peroxidase (HRP) (DAKO, Carpinteria, CA), and 3,3′-diaminobenzidine (DAB) reagents (DAKO). Slides were then counterstained with hematoxylin and mounted with Permount. TUNEL staining was performed using an In Situ Cell Death Detection Kit (Roche Applied Science). For detection of 5-bromo2′-deoxy-uridine (BrdU) incorporation, pregnant mice were intraperitoneally injected with 100 mg/kg BrdU 5 hours prior to embryo harvesting and embryo sections were stained using a BrdU Staining Kit (Zymed, South San Francisco, CA).
In Situ Hybridization
Paraffin sections were prepared as described above and used for in situ hybridizations following a protocol we have described (Stankunas et al., 2008a). Briefly, slides were treated with 0.3% H2O2, washed with tris-buffered saline (TBS), treated with 10 μg/mL Proteinase K, washed with TBS containing 0.1% Tween-20 (TBST), and then incubated overnight with digoxigenin-labeled RNA probe matching the indicated transcript. The slides were then rinsed, treated with an RNase cocktail (Invitrogen), washed, and blocked. The digoxigenin-labeled RNA/DNA was detected using an HRP-conjugated anti-digoxigenin antibody (Roche Applied Science), a signal amplification kit (DAKO) and DAB-based staining. The slides were counterstained with hematoxylin, washed with dH2O, dehydrated, and mounted with Permount prior to imaging.
Valve Elongation Quantification
Matching sections of experimental and littermate control embryos were H&E stained and photographed. Valve leaflet length and three width measurements for each leaflet were recorded in a genotype-blind fashion using Advanced Elements software (Nikon). Length:width ratios were determined by dividing length of each valve by the average of the three width measurements. Ratios were normalized to wildtype ratio mean. A two-tailed t-test was conducted to examine the difference in the means of the length:width ratios between the two groups of interest, and p<0.05 was chosen as the threshold for significance.
Results
Transgenic Approaches to Study VEGF Signaling in Valve Development
To explore the roles of VEGF signaling in valve development, we examined the expression of the VEGFR1 and VEGFR2 receptors at different stages of heart development. Antibody staining against VEGFR2 revealed dynamic changes in its expression in different regions of the heart. At E9.5, prior to the onset of EMT, VEGFR2 protein was found in the endocardium of the common atrium and ventricle (Figure 1A) with distinctly lower levels in the cushion endocardium of both the OFT (Figure S1A) and AVC (Figure 1B). Interestingly, following EMT, VEGFR2 levels became more uniform throughout the endocardium. VEGFR2 expression in cushion endocardium was first apparent in a subset of cells at E10.5 (Figure 1C, 1D) that persisted to at least E12.5 (Figure 1E, 1F, S1B). VEGFR2 transcripts mirrored the protein expression, indicating the tissue-specificity results from transcriptional control (Figure S1C, S1D). Since the VEGFR2 ligand, VEGFA, is a robust inhibitor of EMT, the low levels of VEGFR2 in cushion endocardium at E9.5 may contribute to the unique ability of this region (compared to, for example, ventricular endocardium) to undergo EMT. The later onset of VEGFR2 expression in cushion endocardium could reflect VEGF roles in terminating EMT or modulating the remodeling of these tissues.
Figure 1.
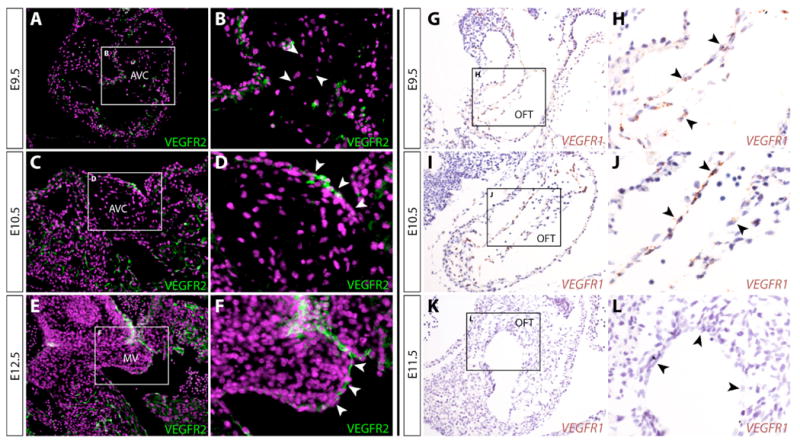
Cushion endocardial cells switch from VEGFR1 to VEGFR2 expression following epithelial-to-mesenchymal transformation (EMT). (A-F) Immunofluorescent staining for VEGFR2 in paraffin sections of atrioventricular canal (AVC) cushions of E9.5 (A, B), E10.5 (C, D), and E12.5 (E, F) wildtype embryos. VEGFR2 expression is shown in green and nuclei (stained with Hoechst) are in purple. (B, D, F) Higher magnification views of panels A, C, and E, respectively. (G-L) In situ hybridization for VEGFR1 transcripts (stained brown with DAB) of E9.5 (G, H), E10.5 (I, J), and E11.5 (K, L) wildtype embryo sections containing the outflow tract (OFT). (H, J, L) Higher magnification views of panels G, I, and K. Counterstain is hematoxylin (blue). AVC: atrioventricular canal cushion. MV: mitral valve leaflet.
VEGFR1 transcripts were expressed in a strikingly complementary pattern to VEGFR2. In the heart, VEGFR1 was restricted to cushion endocardium at E9.5 (OFT, Figure 1G, 1H; AVC, S1E) and E10.5 (OFT, Figure 1I, 1J; AVC, S1F). By E11.5, VEGFR1 levels became undetectable in the heart, including the OFT (Figure 1K, 1L). Since VEGFR1 can act as a decoy receptor for VEGF proteins and oppose VEGFR2-mediated responses (Park et al., 1994), the complementary pattern of VEGFR1 and VEGFR2 expression could reinforce the prevention of VEGF signaling in cushion endocardium until after EMT is complete while facilitating simultaneous signaling in other endocardial cell populations.
To test these possible spatiotemporal roles of VEGF signaling (Figure 2A) in vivo, we employed an inducible “dominant-negative” approach using the tetracycline (tet)-on system (Figure 2B). Transgenic mice (TRE-sFlt) were generated that provide tet-regulated expression of the sFlt protein, an engineered secreted form of VEGFR1 designed to sequester the VEGFR1 ligands, VEGFA, VEGFB, and PlGF (Kuo et al., 2001) (Figure 2C). A second strain (TRE-VEGFR2T) was used that provides expression of VEGFR2T, a modified VEGFR2 protein that remains membrane bound but is incapable of signaling (Carpenter et al., 2005). As such, VEGFR2T blocks signaling mediated by the VEGFR2 ligands, VEGFA, VEGFC, and VEGFD (Figure 2D). The “driver” line employed, Actin-rtTA, provides expression of the reverse tetracycline transactivator (rtTA) in cells where its composite CMV enhancer/β-actin promoter is active (Sarin et al., 2005). Pregnant mice were treated with doxycycline (dox) to drive expression of either sFlt or VEGFR2T during desired periods of embryogenesis (Figure 2B).
Figure 2.
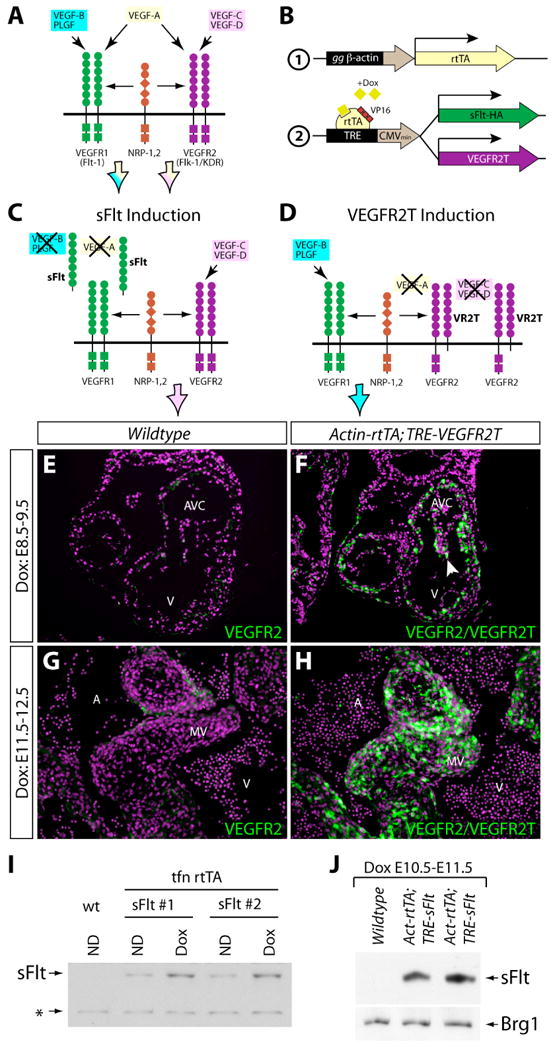
Transgenic mouse approaches used to inducibly inhibit different subsets of VEGF ligands during development. (A) Diagram depicting the specificity of VEGF signaling mediated by different VEGF ligands acting through VEGFR1 or VEGFR2 receptors. (B) Diagram demonstrating the transgenic mouse approach employed for doxycycline (dox)-inducible expression of the VEGF inhibitors sFlt and VEGFR2T (2) using reverse tetracycline-controlled transactivator (rtTA) driven by the gallus gallus (gg) β-actin promoter (1). (C, D) Schematics demonstrating the expected effects on VEGF signaling by the VEGF inhibitors sFlt (C) and VEGFR2T (D). The arrowhead points to cushion endocardial cells. (E-H) Exposure-matched immunofluorescent staining for VEGFR2 and VEGFR2T on heart sections of wildtype (E, G) and Actin-rtTA; TRE-VEGFR2T (F, H) embryos treated with dox from E8.5-9.5 (E, F) or E11.5-12.5 (G, H). VEGFR2 and VEGFR2T are in green, and nuclei are in purple (stained with Hoechst). (I, J) Western blots using an anti-HA antibody to recognize sFlt-HA produced from the TRE-sFlt transgene. (I) sFlt-HA present in conditional media from wildtype (wt) and two TRE-sFlt mouse embryonic fibroblast lines transfected with an Actin-rtTA plasmid and treated with or without dox. ND: no drug. *: background band used as a loading control. (J) sFlt-HA and Brg1 (as a loading control) expression in E11.5 heart protein extracts prepared from wildtype and Actin-rtTA;TRE-sFlt littermate embryos. V: ventricle. AVC: atrioventricular canal cushion. MV: mitral valve leaflet.
We first examined the spatial activity of the Actin-rtTA driver by staining sections of Actin-rtTA;TRE-VEGFR2T dox-treated embryos with an antibody that recognizes both VEGFR2 and VEGFR2T. Embryos treated with dox from E8.5 to E9.5 (Figure 2F) and from E11.5 to E12.5 (Figure 2H) showed very strong expression of VEGFR2T throughout the developing heart, albeit with variation in VEGFR2T levels from cell-to-cell. VEGFR2T expression greatly exceeded VEGFR2 levels, as shown by comparing VEGFR2T staining intensity in experimental embryos with that of endogenous VEGFR2 in littermate controls (Figure 2E and 2G). VEGFR2T induction driven by Actin-rtTA and dox was evident at all stages of development examined (data not shown). The TRE-sFlt mice also produced robust dox-dependent induction. We prepared embryonic fibroblasts from TRE-sFlt mice and transfected them with an rtTA expressing plasmid. We observed a dox-dependent increase in secreted sFlt by western blotting conditioned media using an antibody recognizing the HA-epitope tag engineered on sFlt (Figure 2I). The slight expression of sFlt without added dox may reflect contaminating tetracycline antibiotics in the serum used in the culture media or residual rtTA activity on the TRE promoter in the absence of dox. This latter possibility could contribute to the postnatal lethality of Actin-rtTA;TRE-sFlt mice without dox exposure. In contrast, non-dox treated Actin-rtTA;TRE-VEGFR2T mice were viable and fertile. sFlt protein was also present in protein extracts prepared from hearts of Actin-rtTA;TRE-sFlt embryos exposed to dox from E9.5 to E10.5 (Figure 2J). Further, sFlt induction in mid-gestational embryos for longer than 2 days produced growth retardation and embryonic lethality, likely due to disruption of vascular development. This result demonstrates robust and functional expression of the TRE-sFlt line in utero. Together, Actin-rtTA;TRE-VEGFR2T and Actin-rtTA;TRE-sFlt transgenic mice allow temporal inhibition of VEGF signaling in utero, overcome potential genetic redundancies between VEGF family members, and permit the distinction of roles of VEGF ligand subsets.
VEGFR1 Signaling Contributes to EMT in the OFT
We examined the roles of VEGF signaling in the EMT process by inducing expression of sFlt or VEGFR2T immediately prior to EMT initiation. Actin-rtTA;TRE-sFlt embryos treated with dox beginning at E9.5 and harvested at E10.5 exhibited aberrant EMT in the OFT (Figure 3C, 3D). Instead of producing long, spindle-shaped mesenchymal cells that migrated throughout the cardiac jelly, the Actin-rtTA;TRE-sFlt embryos exhibited a “stacking-up” of cells adjacent to the OFT cushion endocardium. Although these cells had delaminated from the surface epithelium, they appeared histologically intermediate between epithelial and mesenchymal fates, being more rounded in shape, clustered but dissociated from each other, and lacking robust spindles. For descriptive purposes, we termed these cells “endomesenchymal”. We treated Actin-rtTA;TRE-sFlt embryos with dox during different stages of valve development to determine the time window when sFlt expression would produce this phenotype. Endomesenchymal cells within the OFT cushions were not evident upon either an earlier induction of sFlt beginning at E8.75 through E9.75 (Figure 3A, 3B) or a later induction initiated at E10.5 with embryos harvested at E11.5 (Figure 3E, 3F). Additional experiments refined the execution period for sFlt induction to produce endomesenchymal cell accumulation to between E9.0 and E9.75 (Figure 3G). In contrast to the OFT, sFlt induction between neither E8.75 and E9.75 nor E9.5 and E10.5 caused endomesenchymal cells to form in the AVC (data not shown, Figure S2A, S2B). The absence of EMT defects in the AVC upon sFlt induction suggests that peripheral vascular defects caused by sFlt do not indirectly disrupt EMT in the developing heart. Further, these results indicate that sFlt specifically acts in the OFT to disturb cushion mesenchyme formation. Therefore, VEGF signaling is required for proper EMT in the OFT but not AVC during a discrete period when EMT is actively occurring. The signaling may be mediated by VEGFR1, as VEGFR2 was poorly expressed in OFT endocardium at E9.5 and VEGFR2T induction from E8.5-E10.5 (Figure S2E, S2F) did not produce the same phenotype.
Figure 3.
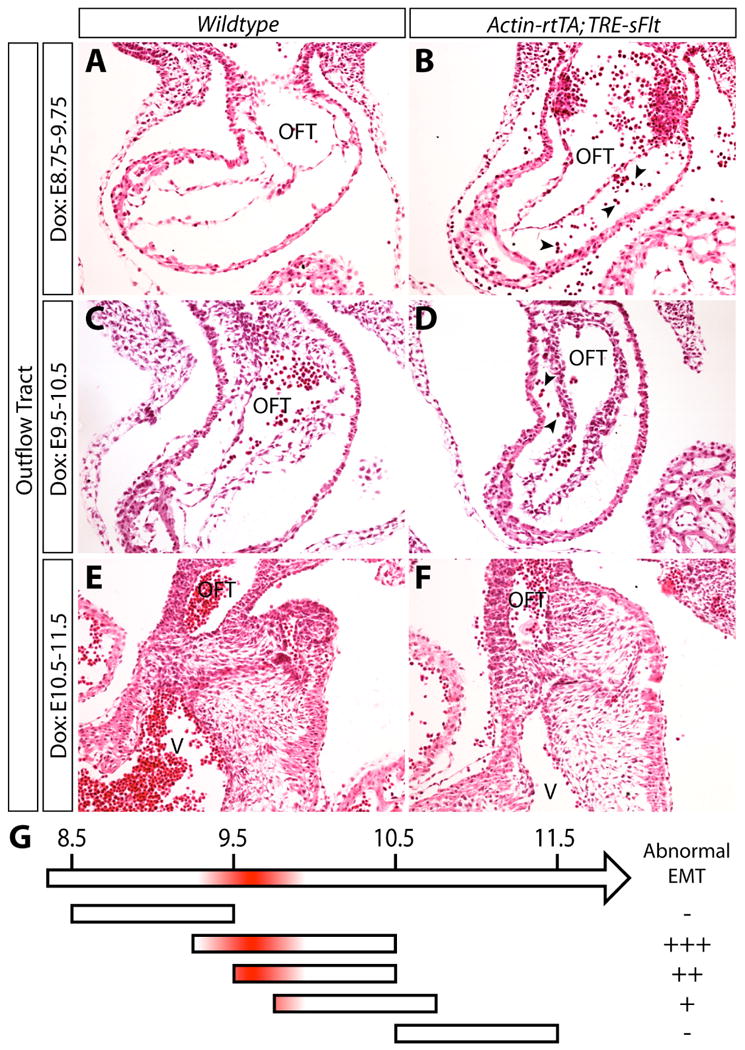
Blocking VEGF signaling using sFlt induction prevents mesenchymal cells from properly invading the cardiac jelly of the outflow tract (OFT) cushions. (A-F) H&E stained sections of the OFT of wildtype (A, C, E) and Actin-rtTA; TRE-sFlt (B, D, F) embryos treated with doxycycline (dox) from E8.75-9.75 (A, B), E9.5-10.5 (C, D), and E10.5-11.5 (E, F). (G) Timeline depicting severity of endocardial-to-mesenchymal transformation (EMT) defects when blocking VEGF signaling using sFlt induction. Shaded red area indicates the time window during which sFlt must be expressed to disrupt EMT. Arrowheads indicate red blood cells. OFT: outflow tract. V: ventricle (right).
The presence of endomesenchymal cells upon sFlt induction was distinct from most other gene-targeted mice exhibiting EMT defects where the endocardium remains as a simple single cell epithelium. Examples include embryos lacking CnB1 or NFATc2/c3/4 (Chang et al., 2004) and those with endothelial-specific deletion of β-catenin (Liebner et al., 2004), SMAD4 (Lan et al., 2007), BMPR1α (Ma et al., 2005; Song et al., 2007), or Alk2 (Wang et al., 2005). We examined whether this phenomena reflected excessive proliferation of endocardial cells, or a failure of differentiated mesenchymal cells to proliferate. We stained sections with a phospho-histone H3 antibody to determine the number of mitotic cells. There was no difference in proliferation of endocardial cells or the intermediate endomesenchymal cells in the E9.5-10.5 dox-treated Actin-rtTA;TRE-sFlt OFT (Figure S3B) when compared to the control OFT (Figure S3A). We also exposed embryos to BrdU in utero and examined its incorporation by antibody staining to monitor the number of cycling cells during the BrdU treatment period. Again, there was no change qualitatively (Figure S3C, S3D) or quantitatively when comparing the percentage of BrdU-positive mesenchymal cells in the OFTs of dox-treated Actin-rtTA;TRE-sFlt embryos and those of their littermate controls (Figure S3E). Therefore, cell proliferation in the OFT was not affected by VEGF inhibition via sFlt induction during this period. Likewise, rates of cell death were unchanged as shown by TUNEL staining (Figure S3F, S3G).
We considered whether the endomesenchymal cells in the OFT cushions of dox-treated Actin-rtTA;TRE-sFlt embryos reflected a population arrested in an intermediate stage of the EMT process. Under this hypothesis, the endocardial cells undergoing EMT would have completed a first differentiation stage and delaminated from the surface epithelium but failed to fully transdifferentiate and migrate through the cardiac jelly. An alternative possibility, which could reflect the specificity of the phenotype to the OFT over the AVC, is that the endomesenchymal cells upon sFlt induction represented abnormal development of neural crest cells (NCCs) migrating into the OFT cushions. To resolve these hypotheses, we stained OFT sections from E9.5-E10.5 dox-treated Actin-rtTA;TRE-sFlt embryos for the endocardial proteins PECAM1 and NFATc1, which are normally rapidly down-regulated in differentiated mesenchymal cells (de la Pompa et al., 1998; Nakajima et al., 1997) and are not expressed in NCCs. In contrast to the mesenchymal cells of littermate control embryos, the endomesenchymal cells of Actin-rtTA;TRE-sFlt dox-treated embryos retained expression of both PECAM1 and NFATc1 (Figure 4A, 4B, 4C, 4D). This result was unique to the OFT and not seen in AVC mesenchyme (data not shown). Interestingly, the OFT endomesenchymal cells also expressed the cushion mesenchymal protein Sox9 (Akiyama et al., 2004; Montero et al., 2002; Rahkonen et al., 2003) (Figure 4E, 4F). Therefore, the endomesenchymal cells displayed molecular features of both endocardial and mesenchymal cells and represent an arrest of the EMT process upon VEGF inhibition rather than a defect in OFT NCCs. The subsequent failure of the endomesenchymal cells to migrate through the cardiac jelly could be caused by their partial epithelial and incomplete mesenchymal properties.
Figure 4.
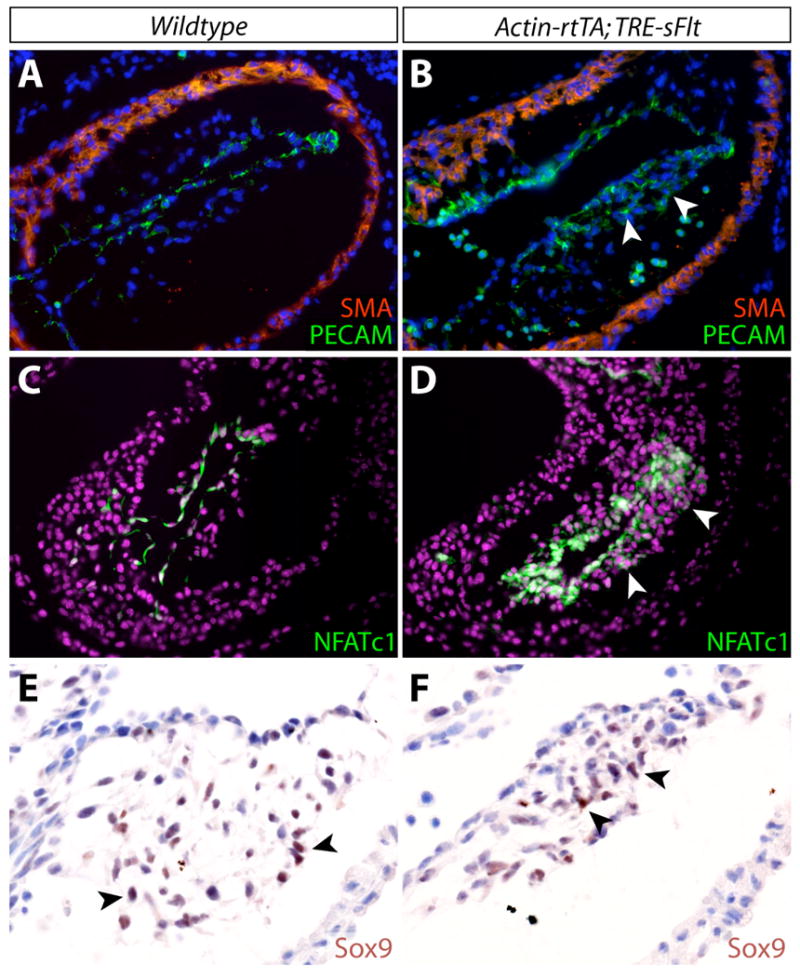
VEGF signaling is required for complete differentiation of outflow tract (OFT) endocardial cells during endocardial-to-mesenchymal transformation (EMT). (A, B) Double immunofluorescent staining for smooth muscle actin (SMA; red) and PECAM (green) in OFT cushions of wildtype (A) and Actin-rtTA; TRE-sFlt (B) E10.5 embryos treated with dox from E9.5-10.5. Nuclei are stained with Hoechst (blue). (C, D) Immunofluorescent staining for NFATc1 (green) in OFT cushions of wildtype (C) and Actin-rtTA; TRE-sFlt (D) E10.5 embryos treated with dox from E9.5-10.5. Nuclei are stained with Hoechst (purple). (E, F) Immunostaining for Sox9 (brown) in OFT cushions of wildtype (E) and Actin-rtTA; TRE-sFlt (F) E10.5 embryos treated with dox from E9.5-10.5. Nuclei are counterstained with hematoxylin (blue). Arrowheads point to cushion mesenchymal cells.
VEGFR2 and miR-126 Regulate Valve Elongation
A second phase of heart valve development follows EMT, when the endocardial cushions undergo morphological changes and elongate to form the valve leaflets. We examined whether VEGF signaling is involved in this aspect of valve development using Actin-rtTA;TRE-VEGFR2T to dox-induce VEGFR2T expression during various periods after E10.5. Expression of VEGFR2T from E10.5 to E13.5 produced mitral valve leaflets that were distinctly shorter and wider than those of wildtype littermates (Figure 5A, 5B). The pulmonic valve leaflets appeared grossly normal (Figure 5E, 5F). To quantitate valve elongation, we carefully matched sections containing the mitral and pulmonic valves from sets of dox-treated Actin-rtTA;TRE-VEGFR2T and littermate control embryos from four independent experiments. We measured the length to width ratio of the leaflets and confirmed the presence of shortened mitral leaflets (Figure 5C, S4) in dox-treated Actin-rtTA;TRE-VEGFR2T embryos. Although not grossly apparent, the pulmonic valve leaflets were also significantly shortened, but with only a minor difference from those of wildtype littermates (Figure 5G, S5). No change was found in the number of cells per area in either valve (Figure 5D, 5H), suggesting that the valve phenotypes reflected morphological defects. We next mapped the time window when VEGFR2T induction produces blunted mitral valves by exposing Actin-rtTA;TRE-VEGFR2T embryos to dox during different periods of embryogenesis. Mitral valve leaflets appeared normal at E12.5 following an E10.5 to E12.5 dox treatment (Figure 5I, 5J), but were blunted when exposed to dox from E11.5 to E13.5 (Figure 5K, 5L). No defects were observed with a dox treatment from E12.5 to E14.5 (data not shown). Therefore, VEGF signaling, as blocked by VEGFR2T expression, is required for valve elongation during a narrow time window centered at E11.5, although the valve blunting phenotype upon VEGFR2 inhibition does not become apparent until the valves have matured to E13.5.
Figure 5.
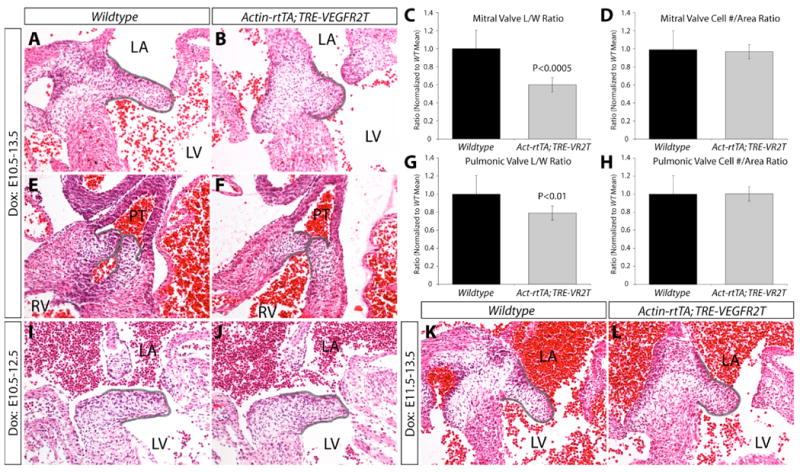
VEGF signaling is required for mitral valve (MV) and, to a lesser extent, pulmonic valve (PV) elongation between embryonic days E10.5 and E13.5. (A, B) H&E stained MV sections from wildtype (A) and Actin-rtTA; TRE-VEGFR2T (B) E13.5 embryos treated with doxycycline (dox) from E10.5-13.5. (C) A quantitative comparison of the length:width ratio of the MV leaflets of wildtype and Actin-rtTA; TRE-VEGFR2T dox-treated E13.5 embryos. n=9 embryos. (D) Measurements of the number of cells per area of MV leaflets of wildtype and Actin-rtTA; TRE-VEGFR2T embryos. n=9 embryos. (E, F) H&E stained sections of PVs from wildtype (E) and Actin-rtTA; TRE-VEGFR2T (F) E13.5 embryos treated with dox from E10.5-13.5. The leaflets are outlined in grey. (G) Morphometric analysis comparing the length: width ratio of PV leaflets of wildtype and Actin-rtTA; TRE-VEGFR2T embryos. n=12 leaflets. (H) Measurements of the number of cells per area of PV leaflets of wildtype and Actin-rtTA; TRE-VEGFR2T embryos. n=12 leaflets. (I-L) H&E stained sections of MVs from wildtype (I, K) and Actin-rtTA; TRE-VEGFR2T (J, L) embryos treated with dox from E10.5-12.5 (I, J) or E11.5-13.5 (K, L). Grey lines are drawn on the H&E staining panels to outline the PV and MV leaflets. LA: left atrium. LV: left ventricle. PT: pulmonary trunk. RV: right ventricle.
In postnatal pulmonic valve endothelial cells, VEGF signaling induces cell proliferation (Johnson et al., 2003). VEGF could likewise regulate localized cell proliferation to drive elongation of embryonic valve leaflets. To examine whether proliferation was defective in Actin-rtTA;TRE-VEGFR2T dox-treated embryos, we stained sections with an anti-phospho-histone H3 antibody to identify mitotic cells. After a dox treatment initiated at E10.5, E12.5 embryos showed only a small number of cells undergoing mitosis (Figure 6A). There was no difference between the experimental and control embryos (Figure 6B, S6A). We also evaluated BrdU incorporation in embryos exposed to BrdU for 5 hours in utero prior to harvesting to examine the number of cells that had undergone DNA replication. There was no difference in BrdU incorporation in either cushion endocardial or mesenchymal cells (Figure 6C, 6D, S6B). Likewise, TUNEL staining revealed no change in the number of apoptotic cells in the mitral valves of Actin-rtTA;TRE-VEGFR2T dox-treated embryos (Figure 6E, 6F, S6C). Therefore, the requirement of VEGF for valve elongation is likely through directing cushion morphogenesis.
Figure 6.
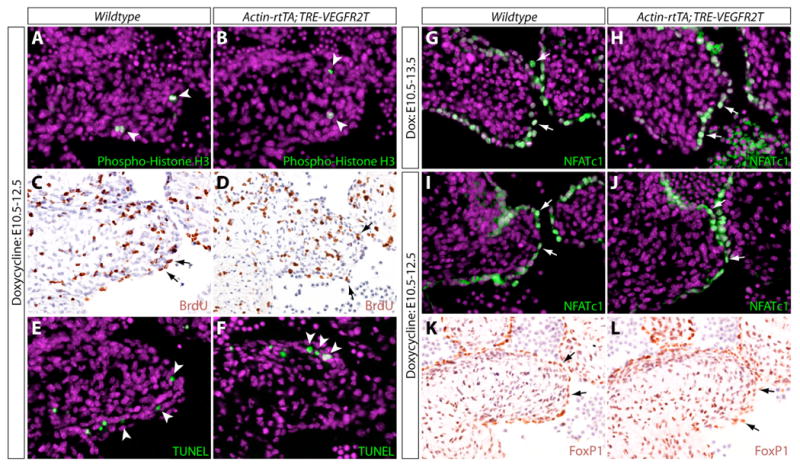
VEGF signaling mediated by VEGFR2 is not required for cell proliferation, cell survival, expression of FoxP1, or nuclear translocation of NFATc1 in endocardial cells of elongating mitral valves (MVs). (A, B) Immunofluorescent staining for phospho-histone H3 (green) in MV sections of wildtype (A) and Actin-rtTA; TRE-VEGFR2T (B) E12.5 embryos treated with doxycycline (dox) from E10.5-12.5. Nuclei are stained with Hoechst (purple). (C, D) Immunostaining for incorporation of BrdU (5 hour exposure in utero; brown) in sections of wildtype (C) and Actin-rtTA; TRE-VEGFR2T (D) E12.5 embryos exposed to dox beginning at E10.5. The nuclear counterstain is hematoxylin (blue). (E, F) TUNEL staining (green) of MVs of wildtype (E) and Actin-rtTA; TRE-VEGFR2T (F) E12.5 dox-treated (E10.5-12.5). Nuclei are stained with Hoechst (purple). (G-J) Immunofluorescent staining for NFATc1 (green) in MVs of wildtype (G, I) and Actin-rtTA; TRE-VEGFR2T (H, J) dox-exposed embryos from E10.5-13.5 (G, H) or E10.5-12.5 (I, J). Nuclei are stained with Hoechst (purple). (K, L) Immunostaining for FoxP1 (brown) in MVs of wildtype (G) and Actin-rtTA; TRE-VEGFR2T (H) E12.5 embryos treated with dox from E10.5-12.5. Nuclei are counterstained using hematoxylin (blue). Arrows point to endocardial cells. Arrowheads point to mesenchymal cells.
NFATc1 is required at E11.5 in endocardial cells for valve elongation (Chang et al., 2004). Previous studies show that VEGF can activate NFATc1 by triggering its nuclear translocation in cultured endothelial cells (Armesilla et al., 1999; Hernandez et al., 2001; Johnson et al., 2003). To test if VEGF regulates the nuclear translocation of NFATc1 in vivo, we examined the subcellular localization of NFATc1 in sections of dox-treated Actin-rtTA;TRE-VEGFR2T mitral valves. We observed no change in the nuclear localization of NFATc1 with either E10.5-E13.5 or E10.5-E12.5 dox treatments despite clear valve elongation defects (Figure 6G, 6H, 6I, 6J). Likewise, there was no difference in the ratio of nuclear versus cytoplasmic NFATc1 staining intensity in mitral valve endocardial cells between wildtype and Actin-rtTA;TRE-VEGFR2T E10.5-E12.5 dox-treated embryos (Figure S6D). Similar results were found in the OFT as well as with either VEGFR2T or sFlt induction during earlier time windows (data not shown). These results suggest that VEGF signaling is not required for NFATc1 nuclear localization in cushion endocardium in vivo. FoxP1, another endocardial transcription factor required for valve elongation (Wang et al., 2004), was also expressed normally in VEGFR2T expressing embryos (Figure 6K, 6L). These observations indicate that VEGF controls heart valve morphogenesis upstream of an undescribed regulatory network.
miR-126, a microRNA expressed specifically in endothelial and endocardial cells (Fish et al., 2008; Kuhnert et al., 2008; Wang et al., 2008), is a known modifier of VEGF signaling. It positively regulates VEGF signaling and other pathways by suppressing the RNA expression of two negative regulators of the VEGF pathway, Pik3r2-encoded p85β subunit of PI3 kinase and Spred1 (Fish et al., 2008; Kuhnert et al., 2008; Liu et al., 2009). miR-126 null embryos show embryonic lethality and severe edema late in embryogenesis with incomplete penetrance (Fish et al., 2008; Kuhnert et al., 2008). We examined whether valve defects resulting from decreased VEGF output could contribute to the embryonic lethality of miR-126 null mice. We analyzed matched sections of the mitral valve leaflets from E13.5 miR-126Δ/Δ and control embryos. A subset of the miR-126Δ/Δ embryos exhibited leaflets that were notably shorter and wider than those of littermate controls (Figure 7A, 7B). We quantitated this valve defect and found a significant decrease in the mean length/width ratio of the medial leaflet. Graphing the distribution showed that the phenotype was incompletely penetrant (Figure 7C) as are other defects and embryonic lethality in miR-126Δ/Δ mice. As with VEGFR2T induction, NFATc1 retained nuclear localization in miR-126Δ/Δ embryos (Figure S7). miR-126Δ/Δ embryos also frequently showed membranous ventricular septal defects and occasionally double outlet of the right ventricle, both phenotypes that likely originate from cushion abnormalities (data not shown). These results show that miR-126 is a component of the endocardial VEGF pathway regulating heart valve morphogenesis. In contrast, the role of VEGF signaling in OFT cushion development during an earlier time window does not require miR-126, underlining how the VEGF pathway dramatically changes in both its components and roles during valve development.
Figure 7.
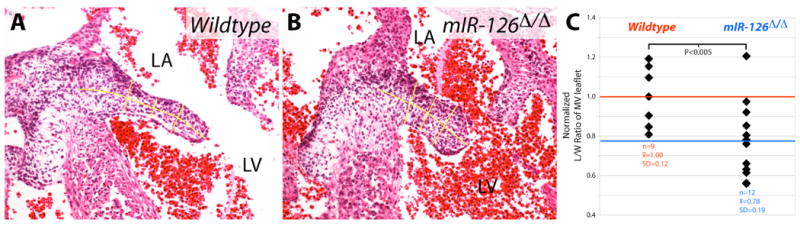
The VEGF-modulating microRNA miR-126 is required for mitral valve (MV) elongation. (A,B) H&E stained MV-containing sections of a wildtype (A) and miR-126Δ/Δ (B) E13.5 embryo. Yellow lines are drawn to show how length and width (averaged) measurements were taken for quantitative analysis. (C) Individual value plot demonstrating the normalized length:width ratios of MVs of wildtype and miR-126Δ/Δ E13.5 embryos. n=9 wildtype MVs; n=12 miR-126Δ/Δ MVs. x̄ = mean length:width ratio. SD = standard deviation. Lines, red for wildtype and blue for miR-126Δ/Δ, indicate the mean values of each group. LA: left atrium. LV: left ventricle.
Discussion
VEGF and Outflow Tract Cushion EMT
Our results demonstrate that VEGF signaling is required during two distinct periods of valve development to first regulate EMT and later valve leaflet elongation. In the OFT but not AVC cushions, inhibition of VEGF signaling results in incomplete cellular differentiation during EMT. In utero induction of sFlt produces delaminated cells in the OFT cushions that lack the mature spindle shape of normal cushion mesenchyme and fail to fully invade the endocardial cushions. Further, these cells express both the endocardial markers NFATc1 and PECAM1 and the mesenchymal protein, Sox9. Our studies therefore define an intermediate population of “endomesenchymal” cells that transiently exists during the transition of endocardial to mesenchymal cells. These results support the proposal that EMT is a multi-step process with distinct and sequential roles for a series of signaling molecules (Armstrong and Bischoff, 2004; Boyer et al., 1999). In the first step, specifically competent endocardial cells are activated, in part, by TGF-β (reviewed in (Yamagishi et al., 2009)) and Notch (Timmerman et al., 2004) signals. TGF-β signaling induces expression of the mesenchymal transcription factors Slug and Snail that subsequently direct changes in surface protein expression to support delamination of the transforming cells. Additional signals, including additional TGF-β proteins and BMP ligands (reviewed in (Armstrong and Bischoff, 2004)) then complete the transdifferentiation process. This final step is characterized by the further loss of endocardial marker expression, including NFATc1, in nascent mesenchymal cells while they acquire a mature, spindle-shaped form and migrate through the cushions. Our results suggest that VEGF signals contribute to these terminal steps of EMT in the OFT cushions.
In contrast to the OFT, the AVC does not require VEGF to complete cushion mesenchymal cell differentiation and instead uses a different set of signals to accomplish this step. Supporting differences between the molecular control of EMT in the OFT and AVC cushions, hypomorphic BMPRII mice show cushion defects only in the OFT (Delot et al., 2003) while endothelial deletion of BMPR1A (Alk3) or activin A receptor, type 1 (Alk2) only disrupts AVC cushion formation (Song et al., 2007; Wang et al., 2005). The sensitivity to VEGF inhibition during OFT but not AVC EMT could be explained by variable cellular origins of the tissues. These differences include the contribution of NCC to the OFT but not AVC cushions (although likely not a direct effect as discussed in the Results section), origin of the cushions from different heart fields, and differential contributions by circulating progenitor cells and/or epicardially derived cells. Regardless, the OFT and AVC cushions have evolved variant strategies, including the use of VEGF signaling, to generate heart valves with distinct morphology and function. These regulatory differences may explain why most patients with congenital valve defects display abnormalities in only one of the two sets of valves, although non-viable pregnancies when fetuses have severe disruptions in both sets of valves also underlies this observation.
Surprisingly, the VEGF-mediated EMT differentiation in the OFT is sensitive to sFlt but not VEGFR2T-mediated inhibition of VEGF signaling. sFlt sequesters VEGF ligands that can bind VEGFR1, thereby blocking a combination of VEGFA, VEGFB, and PlGF-mediated signals. However, VEGFB and PlGF are not essential for EMT in the OFT as null mutations of VEGFB, PlGF or both genes in mice do not cause overt endocardial cushion abnormalities (Bellomo et al., 2000; Carmeliet et al., 2001). Therefore, VEGFA is likely the dominant VEGF ligand that controls EMT in the OFT. However, VEGFA may act redundantly with VEGFB and/or PlGF given that VEGFR2T expression, which sequesters VEGFA but not VEGFB or PlGF, does not reproduce the EMT differentiation defect. This result also points to VEGFR1 rather than VEGFR2 as mediating the VEGF signals during OFT EMT, a model further supported by heightened VEGFR1 levels and reduced VEGFR2 expression in OFT endocardium when EMT is occurring. Additionally, the ventricles of VEGFR1-null embryos display “stacked” or clustered endocardial cells (Fong et al., 1995), a phenotype that resembles the accumulation or stacking of endomesenchymal cells in the OFT caused by sFlt. An active signaling role for VEGFR1 in the developing OFT endocardium is surprising given that deletion of the signaling domain of VEGFR1 in mice does not produce developmental abnormalities (Hiratsuka et al., 1998). Possibly, VEGFR1 partners with low levels of VEGFR2 or other co-receptors, including the Neuropilins (Soker et al., 1998), which directly transmit the VEGF signal intracellularly. Unfortunately, neither VEGFR1 nor VEGFA-null embryos survive long enough to observe EMT phenotypes (Carmeliet et al., 1996; Fong et al., 1995) and the lethal haploinsufficiency of VEGFA (Carmeliet et al., 1996; Ferrara et al., 1996) makes genetic testing of redundant roles with other VEGF family members difficult.
VEGF and Heart Valve Elongation
VEGFA inhibits endocardial cushion EMT and loss of its transcriptional repression blocks EMT, leading to the suggestion that increased VEGFA levels in the endocardial cushion region following EMT may help terminate EMT (Chang et al., 2004; Dor et al., 2001). Further, VEGFA expression may restrict EMT to E9.5 cushion endocardium (Dor et al., 2003). However, our VEGF inhibition studies using VEGFR2T induction did not show an increase in the number of cushion mesenchymal cells or ectopic EMT. This negative result could also be explained by the VEGFR2T not fully inhibiting the appropriate combination of VEGF ligands or that other pathways act redundantly with VEGF signals to terminate EMT. We do demonstrate a new role for VEGF signaling in valve leaflet morphogenesis. Consistent with this, VEGFR2 is expressed in cushion endocardium during the elongation phase of valve morphogenesis, and blocking VEGFR2 signaling by VEGFR2T inhibits valve elongation. It remains to be determined which VEGF family member(s) participate in valve elongation and whether VEGF signals originate from myocardial, mesenchymal, and/or other cells. Ligands that bind VEGFR2 include VEGFA, VEGFC, and VEGFD, all of which are present in the developing heart (Chang et al., 2004; Dor et al., 2001; Lagercrantz et al., 1998; Lavine et al., 2006; Miquerol et al., 1999). Due to VEGFA's early and widespread developmental roles, its temporospatial-controlled deletion is required to test its contributions to valve elongation. On the other hand, VEGFC-/- mice, which exhibit abnormal lymphatic vasculature formation and die late in embryogenesis (Karkkainen et al., 2004), could have un-described valve abnormalities. Similarly, VEGFD-/- mice, which are viable (Baldwin et al., 2005) and do not significantly enhance the VEGFC null lymphatic vasculature phenotype (Haiko et al., 2008), have not been analyzed for valve defects.
Severe disruptions in blood flow can affect heart valve development (Butcher and Markwald, 2007; Hove et al., 2003), suggesting that the valve elongation defects observed in dox-treated Actin-rtTA;TRE-VEGFR2T embryos may be a secondary result of vascular abnormalities. However, our results are most consistent with VEGF signaling acting directly at the AVC cushion endocardium. First, valve defects occurred with a two day dox exposure from E11.5 to E13.5, when the embryos were otherwise indistinguishable from wildtype littermates (data not shown). These observations suggest the absence of a severe vascular or circulatory defect during this window of VEGFR2T induction. Second, cushion mesenchymal cell number, heart size, and heart morphology (with the exception of the heart valves) in E10.5-E13.5 dox-exposed Actin-rtTA;TRE-VEGFR2T embryos were grossly normal (data not shown). In contrast, blood flow disruption caused developmental arrest and multiple cardiac defects in addition to valve abnormalities in zebrafish (Hove et al., 2003). Therefore, our studies indicate that VEGFR2 activity at the AVC cushions directly controls valve leaflet maturation.
The transcription factor NFATc1 is dynamically expressed in developing endocardium and required for elongation of both OFT- and AVC-derived valve leaflets (Chang et al., 2004; de la Pompa et al., 1998; Ranger et al., 1998; Wu et al., 2007). Previous studies show that VEGF is sufficient to activate NFATc1 nuclear translocation in cultured avian embryonic endocardial cells (Combs and Yutzey, 2009) and human pulmonic valve endothelial cells (Johnson et al., 2003). However, our results indicate that VEGF may not be required for NFATc1 nuclear occupancy in vivo or that it cooperates with additional factors. RANKL (Combs and Yutzey, 2009) represents a promising signal to regulate NFATc1 nuclear localization in valve endocardium. We also found no change in FoxP1 expression, another transcription factor required for leaflet extension, or activation of TGF-β signaling, as monitored by anti phospho-SMAD2,3 antibody staining (data not shown). The molecular effectors of VEGF during valve leaflet elongation remain to be defined.
microRNA Control of Valve Maturation
microRNAs (miRNA) represent an emerging and large class of regulatory small RNAs that post-transcriptionally modulate target gene expression. While genetic deletion of microRNA processing enzymes including Dicer (Bernstein et al., 2003; Kanellopoulou et al., 2005) are early embryonic lethal in mice, only a few individual microRNAs have been shown to regulate mammalian organogenesis. In cardiovascular development, miR-1-2 and miR-17-92 null mice exhibit septal defects (Ventura et al., 2008; Zhao et al., 2007) and mice lacking miR-126 have both vascular abnormalities (Kuhnert et al., 2008; Wang et al., 2008) and, as shown here, valve elongation defects. In many cases, miRNAs do not act as on/off switches, but instead modulate signaling output to maintain a tight range of ultimate effector activity (Mourelatos, 2008). miR-126 may have a similar role in controlling VEGF output (and/or additional pathways) in developing endocardial cells. As such, miR-126 null mice could show both variable penetrance and expressivity of valve elongation defects due to stochastic events during development.
Implications for human disease
While VEGF is best known for its roles in regulating vascular formation, our studies and others demonstrate that VEGF controls many different types of biological processes during development. During valve formation, VEGF roles are dynamic, switching from regulating cell differentiation early in the OFT cushions to tissue morphogenesis later in the AVC cushion-derived mitral valve. In other developmental situations, VEGF regulates cell survival and proliferation. The effects of VEGF signaling in a given context depend on the particular ligand-receptor pair utilized and by the combination of participating proteins and regulatory RNAs that modulate signaling output. The apparent complexity and sensitivity of the networks regulated by VEGF during multiple stages of valve development suggest disturbances in VEGF signaling could lead to different degrees of heart valve malformations in humans. Further studies on the pathways interfacing with VEGF during valve formation will produce new candidate molecules whose disruption could contribute to congenital valve defects.
Supplementary Material
Figure 8.
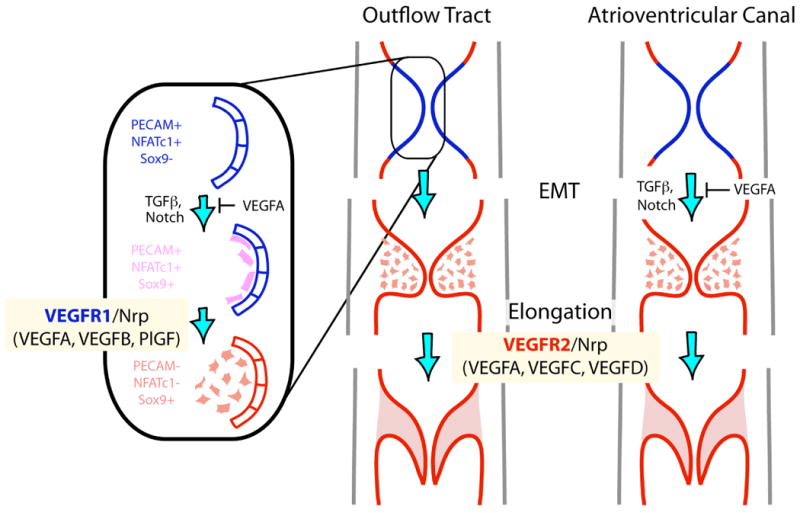
Model of how VEGF signaling roles in valve development switch from a differentiation role during the EMT process involving VEGFR1 and its ligands to a morphogenetic role during valve elongation mediated by VEGFR2 and the microRNA miR-126.
Acknowledgments
We thank Rong Wang for TRE-VEGFR2T mice, Steve Artandi for Actin-rtTA mice and Zhi-Yang Tsun for technical assistance. Bin Zhou and members of the Chang laboratory provided helpful discussions. Lei Chen supported the generation of the TRE-sFlt transgenic mice. K.S. was funded by an American Heart Association (AHA) postdoctoral fellowship, a Pediatric Research Fund pilot award from the Lucille Packard Foundation for Children's Health, and a National Heart, Lung, and Blood Institute (NHLBI) K99/R00 Pathway to Independence award. G.M. was supported by a Howard Hughes Medical Institute Medical Student Research Training Fellowship and an AHA Medical Student Research Award. C.P.C. was supported by grants from the NHLBI, AHA, Baxter Foundation, Children's Heart Foundation, March of Dimes Foundation, Oak Foundation, Office of the University of California (TRDRP), California Institute of Regenerative Medicine, and Stanford Cardiovascular Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci U S A. 2004;101:6502–7. doi: 10.1073/pnas.0401711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armesilla AL, Lorenzo E, Gomez del Arco P, Martinez-Martinez S, Alfranca A, Redondo JM. Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol. 1999;19:2032–43. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–70. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin ME, Halford MM, Roufail S, Williams RA, Hibbs ML, Grail D, Kubo H, Stacker SA, Achen MG. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol. 2005;25:2441–9. doi: 10.1128/MCB.25.6.2441-2449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo D, Headrick JP, Silins GU, Paterson CA, Thomas PS, Gartside M, Mould A, Cahill MM, Tonks ID, Grimmond SM, Townson S, Wells C, Little M, Cummings MC, Hayward NK, Kay GF. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res. 2000;86:E29–35. doi: 10.1161/01.res.86.2.e29. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–7. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208:530–45. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. First of two parts. N Engl J Med. 2000;342:256–63. doi: 10.1056/NEJM200001273420407. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Markwald RR. Valvulogenesis: the moving target. Philos Trans R Soc Lond B Biol Sci. 2007;362:1489–503. doi: 10.1098/rstb.2007.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R. VEGF is crucial for the hepatic vascular development required for lipoprotein uptake. Development. 2005;132:3293–303. doi: 10.1242/dev.01902. [DOI] [PubMed] [Google Scholar]
- Chang CP, Chen L, Crabtree GR. Sonographic staging of the developmental status of mouse embryos in utero. Genesis. 2003;36:7–11. doi: 10.1002/gene.10186. [DOI] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–63. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Chang CP, Stankunas K, Shang C, Kao SC, Twu KY, Cleary ML. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development. 2008;135:3577–86. doi: 10.1242/dev.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs MD, Yutzey KE. VEGF and RANKL regulation of NFATc1 in heart valve development. Circ Res. 2009;105:565–74. doi: 10.1161/CIRCRESAHA.109.196469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–6. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–20. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, Keshet E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–8. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- Dor Y, Klewer SE, McDonald JA, Keshet E, Camenisch TD. VEGF modulates early heart valve formation. Anat Rec. 2003;271A:202–8. doi: 10.1002/ar.a.10026. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–84. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Jr, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A. 2001;98:5780–5. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh JJ, Gerber HP, Ferrara N, Wagner EF. Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development. 2000;127:1445–53. doi: 10.1242/dev.127.7.1445. [DOI] [PubMed] [Google Scholar]
- Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28:4843–50. doi: 10.1128/MCB.02214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–20. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998;95:9349–54. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove JR, Koster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421:172–7. doi: 10.1038/nature01282. [DOI] [PubMed] [Google Scholar]
- Jacobi J, Tam BY, Wu G, Hoffman J, Cooke JP, Kuo CJ. Adenoviral gene transfer with soluble vascular endothelial growth factor receptors impairs angiogenesis and perfusion in a murine model of hindlimb ischemia. Circulation. 2004;110:2424–9. doi: 10.1161/01.CIR.0000145142.85645.EA. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Lee YM, Sander TL, Rabkin E, Schoen FJ, Kaushal S, Bischoff J. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem. 2003;278:1686–92. doi: 10.1074/jbc.M210250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Dev Biol. 1985;112:414–26. doi: 10.1016/0012-1606(85)90414-2. [DOI] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–93. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- Kuo CJ, Farnebo F, Yu EY, Christofferson R, Swearingen RA, Carter R, von Recum HA, Yuan J, Kamihara J, Flynn E, D'Amato R, Folkman J, Mulligan RC. Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc Natl Acad Sci U S A. 2001;98:4605–10. doi: 10.1073/pnas.081615298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagercrantz J, Farnebo F, Larsson C, Tvrdik T, Weber G, Piehl F. A comparative study of the expression patterns for vegf, vegf-b/vrf and vegf-c in the developing and adult mouse. Biochim Biophys Acta. 1998;1398:157–63. doi: 10.1016/s0167-4781(98)00040-2. [DOI] [PubMed] [Google Scholar]
- Lan Y, Liu B, Yao H, Li F, Weng T, Yang G, Li W, Cheng X, Mao N, Yang X. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol. 2007;27:7683–92. doi: 10.1128/MCB.00577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, White AC, Park C, Smith CS, Choi K, Long F, Hui CC, Ornitz DM. Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006;20:1651–66. doi: 10.1101/gad.1411406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166:359–67. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Peng XC, Zheng XL, Wang J, Qin YW. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer. 2009 doi: 10.1016/j.lungcan.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–11. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- Manasek FJ, Reid M, Vinson W, Seyer J, Johnson R. Glycosaminoglycan synthesis by the early embryonic chick heart. Dev Biol. 1973;35:332–48. doi: 10.1016/0012-1606(73)90028-6. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212:307–22. doi: 10.1006/dbio.1999.9355. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–6. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- Montero JA, Giron B, Arrechedera H, Cheng YC, Scotting P, Chimal-Monroy J, Garcia-Porrero JA, Hurle JM. Expression of Sox8, Sox9 and Sox10 in the developing valves and autonomic nerves of the embryonic heart. Mech Dev. 2002;118:199–202. doi: 10.1016/s0925-4773(02)00249-6. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z. Small RNAs: The seeds of silence. Nature. 2008;455:44–5. doi: 10.1038/455044a. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Mironov V, Yamagishi T, Nakamura H, Markwald RR. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor beta3. Dev Dyn. 1997;209:296–309. doi: 10.1002/(SICI)1097-0177(199707)209:3<296::AID-AJA5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646–54. [PubMed] [Google Scholar]
- Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- Rahkonen O, Savontaus M, Abdelwahid E, Vuorio E, Jokinen E. Expression patterns of cartilage collagens and Sox9 during mouse heart development. Histochem Cell Biol. 2003;120:103–10. doi: 10.1007/s00418-003-0549-9. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–90. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–14. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436:1048–52. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–45. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Song L, Fassler R, Mishina Y, Jiao K, Baldwin HS. Essential functions of Alk3 during AV cushion morphogenesis in mouse embryonic hearts. Dev Biol. 2007;301:276–86. doi: 10.1016/j.ydbio.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Gewillig M, Molin DG, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Plaisance S, Vlietinck R, Emanuel B, Gittenberger-de Groot AC, Scambler P, Morrow B, Driscol DA, Moons L, Esguerra CV, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P. VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med. 2003;9:173–82. doi: 10.1038/nm819. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Bayle JH, Gestwicki JE, Lin YM, Wandless TJ, Crabtree GR. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol Cell. 2003;12:1615–24. doi: 10.1016/s1097-2765(03)00491-x. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008a;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Shang C, Twu KY, Kao SC, Jenkins NA, Copeland NG, Sanyal M, Selleri L, Cleary ML, Chang CP. Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ Res. 2008b;103:702–9. doi: 10.1161/CIRCRESAHA.108.175489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–87. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- Wang J, Sridurongrit S, Dudas M, Thomas P, Nagy A, Schneider MD, Epstein JA, Kaartinen V. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol. 2005;286:299–310. doi: 10.1016/j.ydbio.2005.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–71. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kao SC, Barrientos T, Baldwin SH, Olson EN, Crabtree GR, Zhou B, Chang CP. Down syndrome critical region-1 is a transcriptional target of nuclear factor of activated T cells-c1 within the endocardium during heart development. J Biol Chem. 2007;282:30673–9. doi: 10.1074/jbc.M703622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi T, Ando K, Nakamura H. Roles of TGFbeta and BMP during valvuloseptal endocardial cushion formation. Anat Sci Int. 2009;84:77–87. doi: 10.1007/s12565-009-0027-0. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.