

Abstract
Objectives
Group X secretory phospholipase A2 (GX sPLA2) potently hydrolyzes plasma membranes to generate lysophospholipids and free fatty acids and has been implicated in inflammatory diseases including atherosclerosis. Here we identify a novel role for GX sPLA2 in modulating ABCA1 and ABCG1 expression and hence macrophage cholesterol efflux.
Methods and Results
Overexpression or exogenous addition of GX sPLA2 significantly reduced ABCA1 and ABCG1 expression in J774 macrophage-like cells, whereas GX sPLA2 deficiency in mouse peritoneal macrophages (MPMs) was associated with enhanced expression. Altered ABC transporter expression led to reduced cholesterol efflux in GX sPLA2 overexpressing J774 cells, and increased efflux in GX sPLA2-deficient MPMs. Gene regulation was dependent on GX sPLA2 catalytic activity, mimicked by arachidonic acid, abrogated when LXRα/β expression was suppressed, and partially reversed by the LXR agonist T0901317. Reporter assays indicated that GX sPLA2 suppresses the ability of LXR to trans-activate its promoters through a mechanism involving the C-terminal portion of LXR spanning the ligand binding domain.
Conclusions
GX sPLA2 modulates gene expression in macrophages by generating lipolytic products that suppress LXR activation. GX sPLA2 may play a previously unrecognized role in atherosclerotic lipid accumulation by negatively regulating genes critical for cellular cholesterol efflux.
The secretory phospholipase A2 (sPLA2) family represents a group of structurally related calcium-dependent enzymes that hydrolyze glycerophospholipids at the sn-2 position to liberate lysophospholipids and free fatty acids. Ten enzymatically active sPLA2s have been identified in mammals, of which GX sPLA2 exhibits several unique characteristics. This enzyme has the highest affinity for zwitterionic phospholipids including phosphatidylcholine, and hence is the most potent sPLA2 in hydrolyzing the outer leaflet of intact mammalian membranes in vitro1. GX sPLA2 is synthesized as an inactive zymogen that is processed to an active form through the proteolytic removal of an N-terminal propeptide 2. Based on recent studies in genetically manipulated mice, GX sPLA2 appears to play an important role in inflammatory diseases of the lung 3, 4 and in the pathogenesis of myocardial ischemia/reperfusion injury 5. Interestingly, some of the biological effects attributed to GX sPLA2 in vitro have been shown to be independent of its catalytic activity 6.
GX sPLA2 is present in human and mouse atherosclerotic lesions, where it has been suggested to promote atherosclerotic processes through its ability to hydrolyze lipoprotein particles. In the case of LDL, GX sPLA2 hydrolysis generates an altered particle that is avidly taken up by macrophages to form foam cells7. In this study, we report a novel mechanism by which GX sPLA2 may promote cholesterol accumulation in macrophages. Our data indicate that GX sPLA2 suppresses macrophage expression of ABCA1 and ABCG1, transport proteins that reduce cellular lipid content by effluxing excess cholesterol to extracellular acceptors.
Methods
The supplemental information provides a description of RNA extractions, RT-qPCR, immunoblotting, mouse peritoneal macrophage (MPM) isolation, production of gene-targeted mice, and the generation of J774 cells stably overexpressing FLAG-tagged GX sPLA2 and the catalytically inactive mutant H46Q. More detailed methods for cholesterol efflux, arachidonic acid release assay, siRNA suppression, and reporter assays are also provided in the supplemental information section.
Cell treatments
For some experiments, J774 cells were incubated with 0.1 μg/ml mGX sPLA2 8 in complete medium (DMEM medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin) for 8 h prior to analyses. For other experiments, J774 cells were incubated with 10 μM lysophosphatidylcholine (LPC; Sigma, St. Louis, MO) or vehicle (ethanol) in DMEM medium containing 1% fatty acid-free BSA (Sigma, St. Louis, MO) for 20 h. Alternatively, J774 cells were incubated with 10 μM arachidonic acid (AA; Sigma, St. Louis, MO) or vehicle (DMSO) in DMEM medium containing 1% fatty acid-free BSA for 8 h. For indomethacin (1 μM, Sigma, St. Louis, MO) treatments, the drug or vehicle (ethanol) was added to J774 cells either with or without lipopolysaccharide (LPS; 100 ng/ml, Sigma, St. Louis, MO, Cat # L2654) in complete medium for 6 h. At the end of the incubation time, prostaglandin E2 (PGE2) in cell culture media was quantified by EIA (Cayman Chemical Company, Ann Arbor, Michigan) following the manufacturer's protocol.
Arachidonic acid release assay
Arachidonic acid release assay was carried out as described previously9.
Cholesterol efflux assay
Cholesterol efflux to apoAI (Biodesign International, Saco, Maine) and HDL3 was carried out as described previously 10.
Reporter assays
Luciferase activities were analyzed using the Dual-Luciferase Reporter Assay system (Promega, Madison, Wis).
Statistical analyses
Data are expressed as mean ± SEM. Results were analyzed by Student t-test or one-way ANOVA followed by Bonferroni's post-test. Values of P<0.05 were considered statistically significant. Data was tested for normality and equal variance before analysis.
Results
GX sPLA2 reduces macrophage expression of ABCA1 and ABCG1
In studies investigating the role of GX sPLA2 in macrophage lipid metabolism, we made the unexpected finding that MPMs deficient in GX sPLA2 had significantly increased ABCA1 mRNA, and a trend for increased ABCG1, compared to WT MPMs (Fig. 1A). When MPMs from WT mice were treated with 20 μM indoxam, an sPLA2 inhibitor11, ABCA1 and ABCG1 expression increased 2.4 and 1.7-fold, respectively, an effect that did not reach statistical significance (Supplemental Fig. IA). Incubations with recombinant mGX sPLA2 significantly reduced ABCA1 and ABCG1 expression in J774 macrophage-like cells (Fig. 1B) in a manner that was dose-dependent (Supplemental Fig. IB). Cells treated with GX sPLA2 expressed significantly reduced amounts of ABCA1 protein (Fig. 1C). Consistent with results produced by exogenous addition, forced overexpression of GX sPLA2 resulted in significantly reduced ABCA1 and ABCG1 expression in J774 cells (J774-GX cells) compared to control (J774-C cells) (Fig. 1D; ABCG1 data not shown). Treatments with indoxam restored gene expression to normal levels in J774-GX cells, indicating a specific effect of the transgene to modulate gene expression (Fig. 1D).
Figure 1.
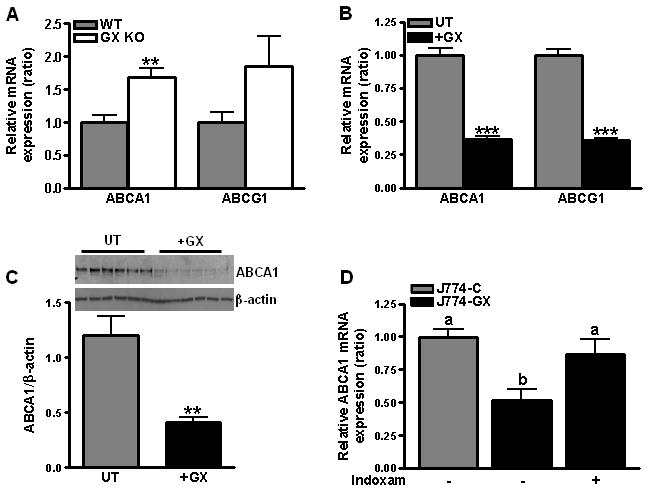
(A) Relative expression of ABCA1 and ABCG1 mRNAs in MPMs from WT and GX KO mice. Data are relative to WT MPM values; **p < 0.01 compared to WT MPMs. (B) J774 cells were treated with or without 0.1 μg/ml GX sPLA2 for 8 hr prior to RT-PCR (n=6); ***, p < 0.001 compared to untreated cells. (C) Western blot analysis of lysates (10 μg protein) from control J774 cells (UT) and cells treated with 0.1 μg/ml recombinant GX sPLA2 (+GX). Results from the densitometric analysis are shown below the blot. **, p < 0.01. (D) Relative expression of ABCA1 mRNA was quantified in untreated J774-C and J774-GX cells and J774-GX cells treated with 20 μM indoxam for 20 h (n = 4) and are representative of 2 experiments. Data that are not significantly different (p ≥ 0.05) are indicated with the same letter.
To directly show that GX sPLA2 hydrolytic activity is required for its regulatory effect, we generated J774 cells stably overexpressing a catalytically inactive mutant (J774-H46Q cells). J774-GX and J774-H46Q cells secreted comparable levels of recombinant protein (Fig. 2A). Culture medium from J774-GX cells exhibited significantly more (4-5-fold) phospholipase activity compared to J774-C or J774-H46Q cells (Fig. 2B). Compared to J774-C cells, expression of ABCA1 and ABCG1 mRNA was significantly reduced in J774-GX cells, but not J774-H46Q cells (Fig. 2C and D). Taken together, our data demonstrate that GX sPLA2 catalytic activity suppresses the expression of ABCA1 and ABCG1 in macrophages.
Figure 2.
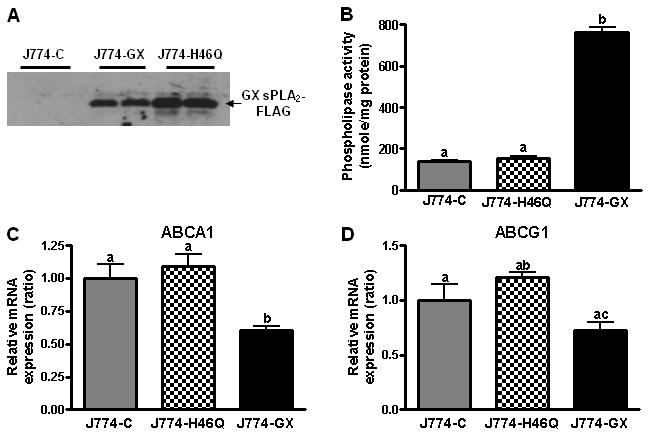
(A) 20 h conditioned media from J774-C, or J774-GX or J774-H46Q was immunoblotted with anti-FLAG antibody. (B) Phospholipase activity in conditioned media from J774-C, J774-H46Q and J774-GX cells. (C and D) Relative expression of ABCA1 and ABCG1 in J774-C, J774-H46Q, and J774-GX cells by RT-PCR (n = 4) and are representative of 3 experiments. Data that are not significantly different (p ≥ 0.05) are indicated with the same letter.
GX sPLA2 reduces macrophage cholesterol efflux
Cholesterol efflux stimulated by apoA1 and HDL was assessed in J774-C, J774-GX, and J774-H46Q cells as well as MPMs isolated from WT and GX KO mice. For these experiments, cells were pre-incubated with the LXR agonist T0901317 to increase the expression of ABCA1 and ABCG1. Such treatments did not abolish the difference in ABCA1 protein expression between J774-C and J774-GX cells (Fig. 3A). J774-GX cells exhibited significantly reduced cholesterol efflux to apoA1 (64% reduction) and HDL (20% reduction) compared to J774-C and J774-H46Q cells (Fig. 3B and 3C), consistent with reduced expression of ABCA1 and ABCG1, respectively. MPMs from GX KO mice showed a modest but significant increase in cholesterol efflux to apoA1 compared to WT cells (Fig. 3D), demonstrating that endogenous GX sPLA2 modulates efflux. The enhanced effluxing capacity of GX KO MPMs was associated with a significant drop in cellular FC content (Fig. 3E).
Figure 3.
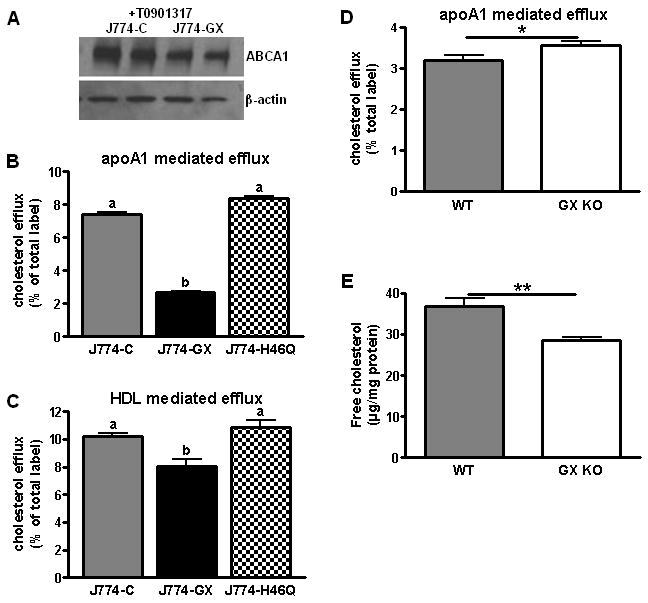
(A) Western blot analysis of cell lysates (10 μg protein) following treatment with 5 μM T0901317 for 12 h. (B) Cellular cholesterol efflux stimulated by 5 h incubations with 10 μg/ml apoA1. (C) Cellular cholesterol efflux stimulated by 5 h incubations with 50 μg/ml HDL3. Data that are not significantly different (p ≥ 0.05) are indicated with the same letter. (D) Cellular cholesterol efflux by MPMs, stimulated by 5 h incubation with 10 μg/ml apoA1. (E) FC content of MPMs isolated from WT and GX KO mice. *p < 0.05; **p < 0.01.
Arachidonic acid suppresses ABCA1 and ABCG1 expression through a COX1/2 independent mechanism
Our data that increased GX sPLA2 catalytic activity leads to suppressed ABCA1 and ABCG1 expression prompted us to investigate which of the enzyme's lipolytic products recapitulate its effects. Whereas treatment of J774 cells with 10 μM lysophosphatidyl choline (LPC) did not significantly alter ABCA1 or ABCG1 mRNA levels (Fig. 4A), the expression of both genes was significantly blunted in cells treated with 10 μM AA (Fig. 4B). J774-GX cells demonstrated significantly increased AA release compared to J774-C cells (Fig. 4C), consistent with GX sPLA2's known ability to hydrolyze intact cell membranes and release AA12. Similarly, exogenous addition of recombinant GX sPLA2 led to significantly enhanced AA release from non-transfected J774 cells (Fig. 4D). On the other hand, deficiency of GX sPLA2 in MPMs was associated with a modest but detectable 16% decrease in AA (data not shown). As expected, the increase in AA release in GX sPLA2 over-expressing cells led to a significant increase in LPS-stimulated PGE2 generation, providing the possibility that GX sPLA2 may alter ABCA1 and ABCG1 expression through the generation of a downstream AA metabolite. To investigate this possibility, J774-C and J774-GX cells were treated with the non-selective COX-1/COX-2 inhibitor indomethacin, which effectively blocked PGE2 production in both cell types upon treatment with LPS (Fig. 4E). However, indomethacin did not normalize ABCA1 or ABCG1 expression in J774-GX cells (Fig. 4F) indicating that GX sPLA2 does not regulate gene expression by amplifying prostanoid synthesis.
Figure 4.
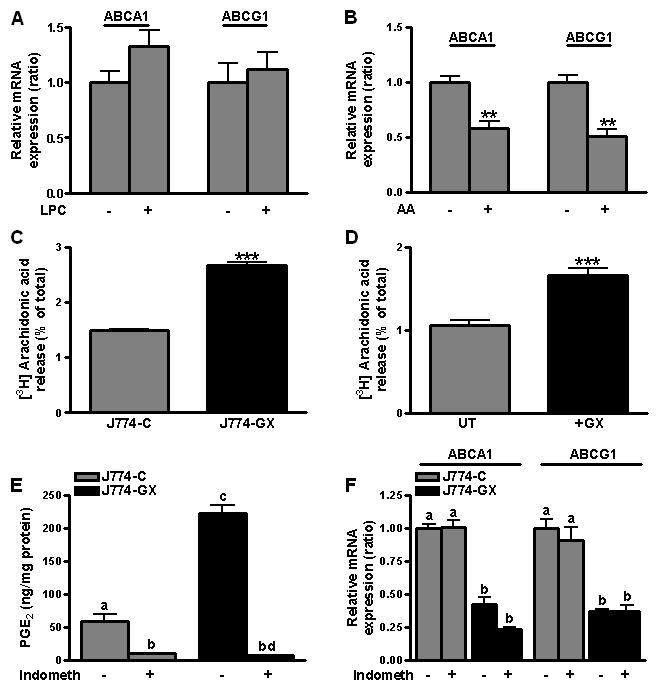
(A) Relative expression of ABCA1 and ABCG1 mRNAs in J774 cells treated for 20 hr with either 10 μM LPC or vehicle (n=6). (B) Relative expression of ABCA1 and ABCG1 mRNAs in J774 cells treated with either 10 μM arachidonic acid (AA) or vehicle for 8 hr (n=4). **p < 0.01 compared to untreated cells. (C) [3H]-AA release during 6 h was quantified and expressed as the percent of total cellular [3H]-AA. (D) [3H]-AA release during 6 h by untreated J774 cells (UT) and J774 cells treated with 0.1 μg/ml GX sPLA2 (+GX) expressed as the percent of total cellular [3H]-AA. ***, p < 0.001. (E) PGE2 in conditioned media of J774-C and J774-GX cells treated for 6 hr with 100 ng/ml LPS in the presence or absence of 1 μM indomethacin (indometh). (F) Relative expression of ABCA1 and ABCG1 mRNAs in J774-C and J774-GX cells incubated with either 1 μM indomethacin or vehicle for 6 hr. Data shown are representative of at least 2 independent experiments. Data that are not significantly different (p ≥ 0.05) are indicated with the same letter.
GX sPLA2 regulates LXR target gene expression
The co-regulation of ABCA1 and ABCG1 suggested the interesting possibility that GX sPLA2 modulates the activity of LXRα/β, known transcriptional activators of these transporters 13, 14. Additional RT-PCR analyses demonstrated that expression of several other known LXR targets (SREBP-1c, LPL, MMP9 and PLTP) was suppressed in J774-GX cells compared to J774-C cells (Supplemental Fig. IIA). Expression of apoE was not altered in J774-GX cells, however, perhaps in line with studies showing that modulation of LXR action may be different amongst diverse target promoters15. GX sPLA2 overexpression was not associated with alterations in transcripts regulated by PPAR-γ (CD36, aP2) or SREBP-2 (LDLR, HMGCoA reductase), or other genes involved in cellular cholesterol metabolism (SR-BI, ACAT, SRA) (Supplemental Fig. IIB).
To investigate whether GX sPLA2 modulates LXR activation, J774-C, J774-H46Q, and J774-GX cells were incubated in the presence or absence of the LXR agonist T0901317. As expected, ABCA1 and ABCG1 expression was significantly induced in J774-C cells after treatment with T0901317 (Fig. 5A,B). Interestingly, the extent of induction was significantly blunted in T0901317-treated J774-GX cells, but not J774-H46Q cells, a finding that was consistent with the protein data presented in Fig. 3A. This reduced induction was not due to altered LXRα or LXRβ mRNA (Supplementary Fig. IIB) or LXRα protein (Supplementary Fig. IIC). We also ruled out the possibility that GX sPLA2 modulates LXR activation by regulating the expression of cholesterol sulfotransferase (SULT2B1), which catabolizes oxysterols that may serve as LXR agonists 16, or Activating Signal Cointegrator-2 (ASC-2), a LXR co-activator 17 (Supplemental Fig. IID). Expression of oxidosqualene cyclase (OSC), which generates 24,25 epoxycholesterol, was significantly reduced in J774-GX cells, possibly as a result of increased cellular FC in J774-GX cells18. However, one might predict that the suppressive effect of GX sPLA2 would be reversible by an LXR agonist if the effect was mediated through decreased OSC expression.
Figure 5.
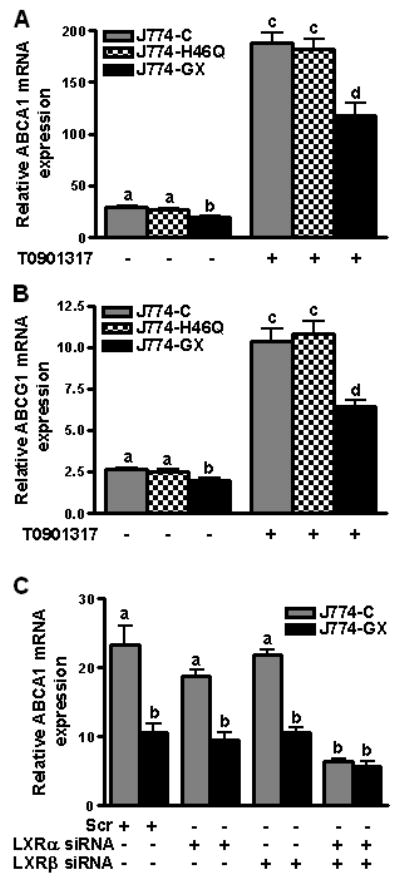
(A and B) J774-C, J774-H46Q, and J774-GX cells were incubated in the presence or absence of 1 μM T0901317 for 20 h as indicated, prior to RT-PCR. Data are means ± SEM (n = 4) and are representative of 3 experiments; values that are not significantly different (p ≥ 0.05) are indicated with the same letter. (C) J774-C and J774-GX cells were transfected with either a control siRNA (scr), siRNA targeting LXRα, siRNA targeting LXRβ or siRNAs targeting both LXRα and LXRβ for 24 h, and then treated with 1 μM T0901317 for an additional 20 h, prior to RT-PCR. Data are means ± SEM (n = 4) and are representative of 2 experiments; Data that are not significantly different (p ≥ 0.05) are indicated with the same letter.
We next determined whether GX sPLA2 decreased LXR target gene expression when LXRα/β was suppressed. Consistent with previous data, ABCA1 mRNA expression was 55% lower in J774-GX cells compared to J774-C cells transfected with a control siRNA (Fig. 5C). This difference in expression of ABCA1 between J774-C and J774-GX did not significantly change when the cells were transfected with siRNA targeting either LXRα or LXRβ, but the difference was totally abolished when cells were cotransfected with siRNAs targeting both LXRα and β. The residual ABCA1 expression in both J774-C and J774-GX cells likely reflects LXR-independent ABCA1 expression 19 and the fact that LXRα/β expression was only partially (60-70% LXRα and 80-90% LXRβ) suppressed (Supplementary Fig. III). These data provide direct evidence that the negative effect of GX sPLA2 on LXR target gene expression is LXRα/β-dependent.
Increased GX sPLA2 activity is linked to suppressed activation of LXR
Taken together, our data suggest that catalytically active GX sPLA2 inhibits the ability of LXR to trans-activate target genes. To investigate whether GX sPLA2 inhibits LXR trans-activation through LXRE, we carried out luciferase reporter assays in HEK293 cells. Compared to control cells, LXRE promoter reporter expression was reduced in cells co-transfected with GX sPLA2 (Fig. 6A). Whereas addition of increasing concentrations of T0901317 dose-dependently increased reporter expression in control cells, the ability of an LXR agonist to activate reporter expression was significantly blunted in cells overexpressing GX sPLA2. Expression of H46Q had no effect on T0901317-induced reporter expression (Fig 6B). Reporter assays carried out in RAW 264.7 macrophages confirmed that GX sPLA2 blunted agonist-induced reporter expression with either T0901317 or 25-OH cholesterol (Supplemental Fig. IV A and B).
Figure 6.
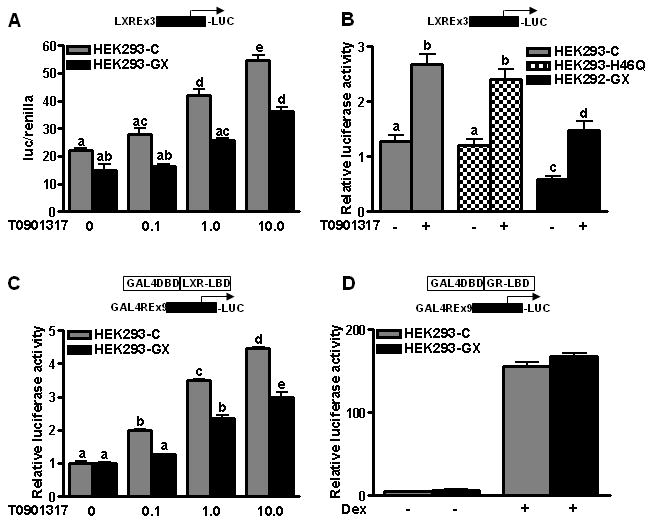
(A) Reporter activity in HEK293 cells transiently transfected with pTK-3xLXRE-Luc reporter construct along with plasmids encoding mLXRα, renilla luciferase, and either mGX sPLA2 expression vector (HEK293-GX) or the corresponding pcDNA3.0 empty expression vector (HEK293-C). Twenty h after transfection, cells were incubated for 24 h with fresh media containing the indicated concentrations of T0901317 (T09).(B) Reporter activity in HEK293 cells transiently transfected with pTK-3xLXRE-Luc reporter construct along with plasmids encoding mLXRα, renilla luciferase, and either mGX sPLA2 expression vector, H46Q (HEK293-H46Q) or the corresponding pcDNA3.0 empty expression vector. Twenty h after transfection, cells were incubated for 24 h with fresh media containing 1 μM T0901317. (C) HEK293 cells were co-transfected with Gal4-driven luciferase reporter construct and an expression vector containing Gal4-DBD fused to LXR-LBD along with GX sPLA2 expression plasmid or control plasmid. After 24 h, the cells were treated with fresh media containing indicated concentrations of T0901317 for 24 h. Data are means ± SEM (n= 4) and are representative of at least 3 independent experiments. (D) The Gal4-driven luciferase reporter construct was cotransfected into HEK293 cells with an expression vector containing Gal4-DBD fused to LBD of glucocorticoid receptor (GR-LBD) and expression plasmid for GX sPLA2 or vector control. After 24 h of transfection, the cells were treated with fresh media containing 10 μM of dexamethasone (DEX) for further 24 h before analyzing the cell lysates for luciferase activity. Data are representative of at least 2 independent experiments. Data that are not significantly different (p ≥ 0.05) are indicated with the same letter.
We next carried out reporter assays utilizing a chimeric construct comprised of the GAL4-DNA binding domain (DBD) fused to the ligand-binding domain (LBD) of mouse LXRα, and a GAL4 response element-driven luciferase reporter construct (Fig. 6C). Reporter activity was significantly reduced when GX sPLA2 was co-expressed, suggesting that the ability of GX sPLA2 to blunt target gene expression is mediated through the LXR LBD. Importantly, GX sPLA2 had no effect on reporter expression driven by a glucocorticoid receptor/GAL4 chimeric construct, demonstrating the specificity of GX sPLA2's inhibitory effect (Fig. 6D).
Accumulating evidence suggests that some LXR agonists can negatively regulate macrophage responses to inflammatory stimuli through trans-repression of NF-κB target genes 20. Thus, it was of interest to determine whether GX sPLA2 modulates the trans-repressive effect of LXR on NF-κB. Murine RAW 264.7 macrophages were transfected with GX sPLA2 along with mLXRα and a NF-κB promoter luciferase construct and then treated with LPS. As reported previously20, NF-κB promoter activation was dampened in control cells treated with LPS in the presence of T0901317 (Supplemental Fig. IVC). Promoter activity was reduced to similar levels in cells transfected with GX sPLA2, indicating that GX sPLA2 does not alter the trans-repressive effects of LXR agonists on NF-κB.
Discussion
ABCA1, and to a lesser extent ABCG1, play key roles in macrophage cholesterol homeostasis by exporting excess cellular FC to extracellular acceptors. LXRs are nuclear hormone receptors that act as intracellular cholesterol sensors by inducing the expression of ABCA1 and ABCG1 in response to cellular FC loading 13, 14. In this study, we identified a novel mechanism for regulating macrophage expression of ABCA1 and ABCG1. We show for the first time that MPMs deficient in GX sPLA2 have significantly increased ABCA1/ABCG1, increased cholesterol efflux, and consequently, decreased cellular FC. This regulatory effect requires GX sPLA2 catalytic activity, is mimicked by the addition of AA to cells, abrogated when LXRα/β is suppressed, and partially reversed by LXR agonists T0901317 and 25-OH cholesterol. Results from luciferase reporter assays support the conclusion that GX sPLA2 suppresses ABCA1 and ABCG1 expression by inhibiting the ability of LXR to trans-activate target genes.
GX sPLA2 is expressed in a number of different tissues including brain, spleen, lung, thymus, intestine, uterus and testis 2, 21, where it has the potential to modulate LXR signaling. We have recently determined that GX sPLA2 is expressed in the adrenal gland, where it regulates steroidogenesis by modulating the expression of steroidogenic acute regulatory protein (StAR) 22, a known LXR target 23,. We also recently reported that hydrolytic products generated by GX sPLA2 negatively regulate adipogenesis, possibly by suppressing LXR activation 24. Given its potent ability to hydrolyze cell membranes and its potential role in multiple metabolic processes, it is important to consider how GX sPLA2 may be regulated in vivo. Studies in vitro implicate the M-type sPLA2 receptor in mediating uptake of extracellular GX sPLA2; however, RT-qPCR analysis of J774 cells and MPMs suggest this receptor is not expressed in macrophages (data not shown). Unlike most members of the sPLA2 family, GX sPLA2 is expressed in an inactive form requiring removal of an N-terminal propeptide for catalytic activity 2. Recent data from the analysis of transgenic mice support the conclusion that GX sPLA2 activity is under tight regulation, and suggest the interesting possibility that proteolytic activation may occur during inflammation 25. Thus, GX sPLA2 may play a key role in coordinating metabolic responses in multiple cell types during inflammation.
Evidence suggests that GX sPLA2 induces some biological effects through a mechanism that is independent of its phospholipase activity 6. Our data that GX sPLA2 but not the inactive mutant H46Q modulates LXR target gene expression provides strong evidence that phospholipid hydrolysis is required for its suppressive effect. This conclusion is further supported by our finding that indoxam (a specific sPLA2 inhibitor) blocked GX sPLA2's effect.
Our data is consistent with the possibility that GX sPLA2 regulates ABCA1 and ABCG1 through the generation of AA. Forced overexpression or exogenous addition of GX sPLA2 resulted in significantly enhanced AA release in J774 cells, whereas GX sPLA2 deficiency in mouse peritoneal macrophages was associated with a 16% reduction in AA release. As expected, accompanying the enhanced AA release in J774-GX cells was a significant increase in LPS-stimulated PGE2 generation. However, we ruled out a role for prostanoids, since the nonselective COX-1/2 inhibitor indomethacin failed to normalize ABC transporter expression in J774 macrophages overexpressing GX sPLA2. According to several reports, polyunsaturated fatty acids including AA act as endogenous LXR antagonists, although the mechanism is unclear. AA has been shown to suppress trans-activation by interacting with the LXR LBD to inhibit LXR/RXR binding to LXREs26, 27. Gel shift mobility and LBD activation assays indicate that PUFAs compete with LXR agonists to block LXR activation 26. PUFAs have also been shown to inhibit LXR function through the activation of PPARα and PPARγ, which in turn reduces the formation of LXR/RXR heterodimers28. Our data demonstrate that GX sPLA2 suppresses LXRE promoter activity through a mechanism involving the C-terminal portion of LXR that spans the LBD. However, the ability of GX sPLA2 to suppress LXR activation as assessed in luciferase reporter assays was not completely reversed by T0901317, suggesting that hydrolytic products generated by GX sPLA2 do not act in a manner that involves direct competition for agonist binding. Nevertheless, the effect of GX sPLA2 was specific to LXR LBD, as there was no suppression of trans-activation when the LXR LBD was replaced by the glucocorticoid receptor LBD. In addition to ligand-binding, the LXR LBD mediates the interaction with corepressors and coactivators29, providing the possibility that GX sPLA2 modulates LXR activation through a mechanism that does not involve competition with agonist.
In addition to playing a central role in macrophage cholesterol homeostasis, considerable evidence now suggests that upon activation by some ligands, LXRs may also regulate inflammatory responses by suppressing NF-κB-mediated gene induction 30. However, our finding that the LXR agonist T0901317 suppresses the activation of a NF-κB promoter construct in response to LPS to a similar extent in J774-C and J774-GX cells suggests that GX sPLA2 overexpression does not interfere with LXR trans-repression of NF-κB. This finding is in keeping with other studies demonstrating that not all LXR ligands known to upregulate ABCA1 transcription are capable of repressing inflammatory gene expression31. It was interesting to note that in the absence of LXR ligand, NF-κB activation induced by LPS was significantly enhanced in macrophages over-expressing GX sPLA2. Although the molecular basis for this effect requires further investigation, GX sPLA2 has been previously implicated in both positive and negative regulation of inflammatory responses3, 6.
The possibility that sPLA2 can also provide lipid mediators (FFAs, prostaglandins) that serve as ligands for PPARs cannot be ruled out; however, evidence that this occurs is currently controversial 32-34. We found no evidence that CD36 or aP2, known PPAR-γ targets, was regulated by GX sPLA2. What remains to be investigated is whether other members of the sPLA2 family can also regulate LXR target gene expression, or if this a unique property of GX sPLA2 due to its potent hydrolytic activity. Interestingly, a suppressive effect of cPLA2 –mediated AA generation on LXR target gene expression was recently reported in primary mouse aortic smooth muscle cells 35. This finding and our current study highlight a new mechanism for regulating gene expression involving hydrolytic products generated by cellular phospholipase A2's.
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance provided by Kathy Forrest. This paper is the result of work supported with resources and the use of facilities at the Lexington VAMC.
Source of Funding: These studies were supported by P01 HL086670 (NRW) and R37 HL036 235 (MHG).
References
- 1.Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 2.Cupillard L, Koumanov K, Mattei MG, Lazdunski M, Lambeau G. Cloning, Chromosomal mapping, and expression of a novel human secretory phospholipase A2. J Biol Chem. 1997;272:15745–15752. doi: 10.1074/jbc.272.25.15745. [DOI] [PubMed] [Google Scholar]
- 3.Curfs DM, Ghesquiere SA, Vergouwe MN, van der Made I, Gijbels MJ, Greaves DR, Verbeek JS, Hofker MH, de Winther MP. Macrophage secretory phospholipase A2 group X enhances anti-inflammatory responses, promotes lipid accumulation, and contributes to aberrant lung pathology. J Biol Chem. 2008;283:21640–21648. doi: 10.1074/jbc.M710584200. [DOI] [PubMed] [Google Scholar]
- 4.Henderson WR, Jr, Chi EY, Bollinger JG, Tien Yt, Ye X, Castelli L, Rubtsov YP, Singer AG, Chiang GKS, Nevalainen T, Rudensky AY, Gelb MH. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med. 2007;204:865–877. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujioka D, Saito Y, Kobayashi T, Yano T, Tezuka H, Ishimoto Y, Suzuki N, Yokota Y, Nakamura T, Obata JE, Kanazawa M, Kawabata K, Hanasaki K, Kugiyama K. Reduction in myocardial ischemia/reperfusion injury in group X secretory phospholipase A2-deficient mice. Circulation. 2008;117:2977–2985. doi: 10.1161/CIRCULATIONAHA.107.743997. [DOI] [PubMed] [Google Scholar]
- 6.Granata F, Petraroli A, Boilard E, Bezzine S, Bollinger J, Del Vecchio L, Gelb MH, Lambeau G, Marone G, Triggiani M. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J Immunol. 2005;174:464–474. doi: 10.4049/jimmunol.174.1.464. [DOI] [PubMed] [Google Scholar]
- 7.Hanasaki K, Yamada K, Yamamoto S, Ishimoto Y, Saiga A, Ono T, Ikeda M, Notoya M, Kamitani S, Arita H. Potent Modification of Low Density Lipoprotein by Group X Secretory Phospholipase A2 Is Linked to Macrophage Foam Cell Formation. J Biol Chem. 2002;277:29116–29124. doi: 10.1074/jbc.M202867200. [DOI] [PubMed] [Google Scholar]
- 8.Rouault M, Le Calvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Bollinger J, Gelb MH, Lambeau G. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry. 2007;46:1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- 9.Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-{alpha} J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 10.Francis GA, Knopp RH, Oram JF. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier Disease. J Clin Invest. 1995;96:78–87. doi: 10.1172/JCI118082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boilard E, Rouault M, Surrel F, Le Calvez C, Bezzine S, Singer A, Gelb MH, Lambeau G. Secreted phospholipase A2 inhibitors are also potent blockers of binding to the M-type receptor. Biochemistry. 2006;45:13203–13218. doi: 10.1021/bi061376d. [DOI] [PubMed] [Google Scholar]
- 12.Bezzine S, Koduri RS, Valentin E, Murakami M, Kudo I, Ghomashchi F, Sadilek M, Lambeau G, Gelb MH. Exogenously added human group X Secreted phospholipase A2 but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J Biol Chem. 2000;275:3179–3191. doi: 10.1074/jbc.275.5.3179. [DOI] [PubMed] [Google Scholar]
- 13.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent Transactivation of the ABC1 Promoter by the Liver X Receptor/Retinoid X Receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 14.Venkateswaran A, Repa JJ, Lobaccaro JMA, Bronson A, Mangelsdorf DJ, Edwards PA. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. J Biol Chem. 2000;275:14700–14707. doi: 10.1074/jbc.275.19.14700. [DOI] [PubMed] [Google Scholar]
- 15.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuda H, Lee YC, Shimizu C, Javitt NB, Strott CA. Mutational analysis of human hydroxysteroid sulfotransferase SULT2B1 isoforms reveals that exon 1B of the SULT2B1 gene produces cholesterol sulfotransferase, whereas exon 1A yields pregnenolone sulfotransferase. J Biol Chem. 2002;277:36161–36166. doi: 10.1074/jbc.M207165200. [DOI] [PubMed] [Google Scholar]
- 17.Mahajan MA, Samuels HH. Nuclear hormone receptor coregulator: role in hormone action, metabolism, growth, and development. Endocr Rev. 2005;26:583–597. doi: 10.1210/er.2004-0012. [DOI] [PubMed] [Google Scholar]
- 18.Dang H, Liu Y, Pang W, Li C, Wang N, Shyy JYJ, Zhu Y. Suppression of 2,3-Oxidosqualene cyclase by high fat diet contributes to liver X receptor-α-mediated improvement of hepatic lipid profile. J Biol Chem. 2009;284:6218–6226. doi: 10.1074/jbc.M803702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Repa JJ, Turley SD, Lobaccaro JMA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of Absorption and ABC1-Mediated Efflux of Cholesterol by RXR Heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 20.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X Receptor-dependent Repression of Matrix Metalloproteinase-9 Expression in Macrophages. J Biol Chem. 2003;278:10443–10449. doi: 10.1074/jbc.M213071200. [DOI] [PubMed] [Google Scholar]
- 21.Morioka Y, Saiga A, Yokota Y, Suzuki N, Ikeda M, Ono T, Nakano K, Fujii N, Ishizaki J, Arita H, Hanasaki K. Mouse Group X secretory phospholipase A2 induces a potent release of arachidonic acid from spleen cells and acts as a ligand for the phospholipase A2 receptor. Arch Biochem Biophys. 2000;381:31–42. doi: 10.1006/abbi.2000.1977. [DOI] [PubMed] [Google Scholar]
- 22.Shridas P, Bailey WM, Boyanovsky BB, Oslund RC, Gelb MH, Webb NR. Group X secretory phospholipase A2 regulates the expression of steroidogenic acute regulatory protein (StAR) in mouse adrenals. J Biol Chem. 2010;285:20031–20039. doi: 10.1074/jbc.M109.090423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummins CL, Volle DH, Zhang Y, McDonald JG, Sion Bt, Lefrançois-Martinez AM, Caira Fo, Veyssière G, Mangelsdorf DJ, Lobaccaro JMA. Liver X receptors regulate adrenal cholesterol balance. J Clin Invest. 2006;116:1902–1912. doi: 10.1172/JCI28400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Shridas P, Forrest K, Bailey W, Webb NR. Group X secretory phospholipase A2 negatively regulates adipogenesis in murine models. FASEB J. 2010 Jun 28; doi: 10.1096/fj.10-154716. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohtsuki M, Taketomi Y, Arata S, Masuda S, Ishikawa Y, Ishii T, Takanezawa Y, Aoki J, Arai H, Yamamoto K, Kudo I, Murakami M. Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J Biol Chem. 2006;281:36420–36433. doi: 10.1074/jbc.M607975200. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, Nakakuki M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Takahashi A, Sone H, Osuga Ji, Gotoda T, Ishibashi S, Yamada N. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem. 2002;277:1705–1711. doi: 10.1074/jbc.M105711200. [DOI] [PubMed] [Google Scholar]
- 27.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98:6027–6032. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshikawa T, Ide T, Shimano H, Yahagi N, Amemiya-Kudo M, Matsuzaka T, Yatoh S, Kitamine T, Okazaki H, Tamura Y, Sekiya M, Takahashi A, Hasty AH, Sato R, Sone H, Osuga Ji, Ishibashi S, Yamada N. Cross-talk between peroxisome proliferator-activated receptor (PPAR) {alpha} and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through Inhibition of LXR signaling. Mol Endocrinol. 2003;17:1240–1254. doi: 10.1210/me.2002-0190. [DOI] [PubMed] [Google Scholar]
- 29.Hu X, Li S, Wu J, Xia C, Lala DS. Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol. 2003;17:1019–1026. doi: 10.1210/me.2002-0399. [DOI] [PubMed] [Google Scholar]
- 30.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 31.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawliczak R, Han C, Huang XL, Demetris AJ, Shelhamer JH, Wu T. 85-kDa cytosolic phospholipase A2 mediates peroxisome proliferator-activated receptor gamma activation in human lung epithelial cells. J Biol Chem. 2002;277:33153–33163. doi: 10.1074/jbc.M200246200. [DOI] [PubMed] [Google Scholar]
- 33.Ziouzenkova O, Perrey S, Asatryan L, Hwang J, MacNaul KL, Moller DE, Rader DJ, Sevanian A, Zechner R, Hoefler G, Plutzky J. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: Evidence for an antiinflammatory role for lipoprotein lipase. Proc Natl Acad Sci U S A. 2003;100:2730–2735. doi: 10.1073/pnas.0538015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Namgaladze D, Morbitzer D, von Knethen A, Brune B. Phospholipase A2-modified low-density lipoprotein activates macrophage peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2009;30:313–320. doi: 10.1161/ATVBAHA.109.199232. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L, Choi HY, Li WP, Xu F, Herz J. LRP1 Controls cPLA2 phosphorylation, ABCA1 expression and cellular cholesterol export. PLoS ONE. 2009;4:e6853. doi: 10.1371/journal.pone.0006853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.