

Abstract

The formaldehyde-inhibited Mo(V) state of xanthine oxidase (I) has been studied for four decades, yet it has not proven possible to distinguish unequivocally among the several structures proposed for this form. The uniquely large isotropic hyperfine coupling for 13C from CH2O led to the intriguing suggestion of a direct Mo-C bond for the active site of I. This suggestion was supported by the recent crystal structures of glycol- and glycerol-inhibited forms of aldehyde oxidoreductase (AOR), a member of the xanthine oxidase family. 1,2H-ENDOR spectra of I(C1,2H2O) in H2O/D2O buffer now unambiguously reveal that the active-site structure for I contains a CH2O adduct of Mo(V) in the form of a four-membered ring with S and O linking the C to Mo, and rule out a direct Mo-C bond. DFT computations are consistent with this conclusion. We interpret the large 13C coupling as resulting from a 'transannular hyperfine interaction'.
Xanthine oxidase is a molybdo-enzyme that catalyses the oxidative hydroxylation of a variety of heterocyclic and aldehyde substrates, including the physiological substrates hypoxanthine and xanthine.1 Of the large number of EPR-active Mo(V) intermediates exhibited by this enzyme,2 one of the most intriguing is the CH2O inhibited Mo(V) form (I) first described by Bray and coworkers.3 Its most remarkable feature, revealed by Howes et al., is the presence of a carbon derived from CH2O that EPR and ENDOR spectroscopies show to have a uniquely large 13C isotropic hyperfine coupling, aiso (13C) ~ 43.0 MHz.4,5 This value contrasts with the five-fold smaller coupling for the Mo-O- 13C of the “2-hydroxy-6-methyl-purine (HMP) very rapid” form (aiso = 7.9 MHz).6 Although I has been studied for 40 years, it has not proven possible to distinguish unequivocally among the viable candidates for its structure (Scheme 1).5,7–13
Scheme 1.
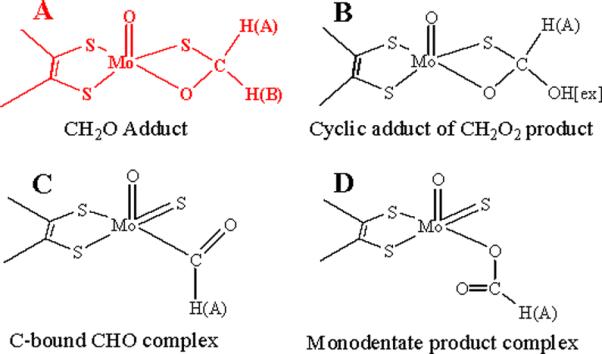
The large isotropic 13C hyperfine coupling for I led Howes et al. to the intriguing suggestion that it contains a CHO fragment with a direct Mo-C bond, Scheme 1, C.4,5 This suggestion recently received support from the crystal structures of glycol and glycerol inhibited forms of aldehyde oxidoreductase (AOR), a member of the xanthine oxidase family.7 These structures exhibit Mo-C bond distances of 2.36 Å and 2.72 Å, respectively, the former in particular being suggestive of direct Mo-C bonding interactions. In contrast, analysis of the smaller 13C coupling for a substrate-derived species bound to Mo(V) of the “very rapid” state of xanthine oxidase indicated there was no direct Mo-C bond,6 and this was confirmed by recent crystal structures for that intermediate.14,15
We here report that 1,2H-ENDOR spectra of I(C1,2H2O) prepared16 in H2O/D2O buffer rule out all models proposed for the active-site structure of I (Scheme 1) except for model A, a four-membered cyclic adduct of CH2O with S and O linking the C to Mo. DFT calculations are consistent with the ENDOR finding that A represents the structure for the active site of I, and not the direct Mo-C bond of C.
The X-band EPR spectrum of I(12,13C1H2O) in H2O shows a doublet splitting from the 13C-nucleus, with each line further split into a doublet from a single proton that derives from CH2O.4 The 35 GHz echo-detected EPR spectrum of I(12,13C1H2O) in H2O show the 13C doublet from 13C1H2O, but the 1H splitting is not observed, Fig S1.17,18 The EPR spectrum of I(12,13C1H2O) further shows hyperfine splitting from 95,97Mo (natural abundance: 15.9% 95Mo; 9.6% 97Mo). The g1 and g3 splittings are observable in both X and Q band spectra; those associated with g2 are only resolved at Q-band (Fig S1). Simulations of the EPR spectra gave principal g-values and A for the 13C of CH2O that agree with those of Howes et al.: g = [1.988, 1.974, 1.948], A(13C) = [51.5, 40, 40] MHz, aiso = 43.8 MHz.
Fig 1 presents 35 GHz Davies 1H and 95,97Mo (upper), and Mims 2H (lower) ENDOR spectra collected for I(12C1,2H2O) in H2O/D2O near g2, the magnetic field corresponding to the maximum EPR intensity. The Davies spectra of I(C1H2O) in H2O and D2O are essentially the same (A), both exhibiting a 1H doublet centered at the 1H-nuclear Larmor frequency and split by A ≈ 13 MHz. This doublet is absent in the spectrum of I(C2H2O) in H2O (B) and thus is associated with a proton, 1HA, derived from CH2O. The 1H doublet in Fig 1 rides on 95,97Mo-ENDOR signals and is more clearly seen upon subtraction of the spectrum of I(C2H2O, B) from that of I(C1H2O, A), spectrum A–B. The 1HA doublet is replaced by a corresponding 2HA signal when C2H2O is the substrate. Davies 2H-ENDOR spectra of I(C2H2O) in H2O exhibit a 2HA doublet without resolved quadrupole splitting (not shown). The hyperfine interaction, A(2HA) corresponds to a smaller value of A(1HA) than that seen directly in the 1H-ENDOR spectrum of I(C1H2O) in H2O/D2O because of a substantial isotope effect on the hyperfine couplings.19,20
Figure 1.
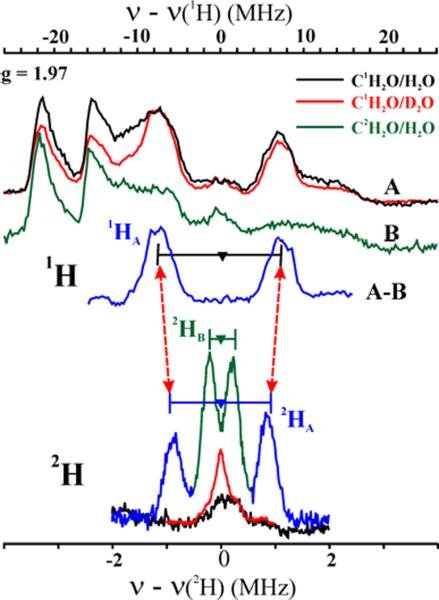
35 GHz (Upper) Davies 1H and 95,97Mo (A, B), ENDOR spectra of I(C1,2H2O) in H2 and D2O buffer. (Lower) Mims 2H spectra of I(C2H2O) in H2O (green/blue), I(C1H2O) in H2O and D2O buffer. Horizontal bars indicate hyperfine splittings for 1,2HA,B non-exchangeable protons. Conditions: g = 1.97; T = 2 K. Davies: π-pulse = 80 ns, τ = 600 ns, repetition time = 50 ms, 34.84 GHz; Mims, 2H: π/2 pulse = 50 ns, τ = 800 ns, repetition time = 50 ms, 34.87 GHz.
To test for both exchangeable and non-exchangeable 2H signal(s) with smaller hyperfine couplings than 1,2HA, Mims 2H-ENDOR spectra were collected for I(C1H2O) in D2O and I(C2H2O) in H2O at g2 (Fig 1, lower). The nearly featureless 2H spectrum of I(C1H2O) in D2O shows clearly that there are no exchangeable protons in the active site of I. However, the 2H spectrum of I(C2H2O) in H2O shows two doublets from deuterons derived from the C2H2O (Fig 1). The larger coupling corresponds to 2HA observed in the Davies 1H-ENDOR spectra of I(C1H2O) in H2O/D2O, again with the isotope effect on the hyperfine coupling. The second doublet in the Mims spectrum for I(C2H2O) also does not exchange in H2O buffer, and thus corresponds to the second C–H deuteron introduced with the C2H2O, denoted 2HB.
To confirm that the Mims 2H spectrum is not significantly distorted by suppression `holes' associated with this technique21 and to determine the 2H hyperfine and quadrupolar tensors, we collected a complete 2-D field-frequency 2H-ENDOR pattern of spectra acquired at numerous magnetic fields across the EPR envelope and simulated this pattern with inclusion of the suppression effects (Fig S2).22 At magnetic fields near g1, the spectra show the 2HA and 2HB doublets centered at 2H-Larmor frequency and split by A ~ 1.8 MHz and 0.4 MHz respectively. The 2H-ENDOR pattern does not change significantly as the field is moved towards g2 and g3, except in intensity, indicating that the hyperfine couplings of 2HA and 2HB are largely isotropic. The 2-D pattern is well simulated by the 1:1 summation of simulations of2HA and 2HB, with, A(2HA) = [1.8, 1.8, 1.9] MHz; A(2HB) = [0.44, 0.4, 0.39] MHz. Neither 2H exhibits quadrupolar splitting (I = 1) because of the large ENDOR line widths. As noted above for the g2 spectrum, the tensor obtained from simulations of the 2HA-ENDOR pattern only approximately matches that obtained by fitting a 2-D 1H Davies ENDOR pattern for 1HA because of an isotope effect on the hyperfine couplings.19,20
The observation that both aldehydic protons of CH2O are non-exchangeably hyperfine-coupled to the Mo(V) of I rules out all candidates in Scheme 1 for the active-site structure of I except the cyclic CH2O adduct, A.
DFT calculations are consistent with experiment in showing a large aiso for the 13C. of Model A. This model yielded hyperfine tensors for 13C, 2HA and 2HB of CH2O that all are in satisfactory agreement with the experimentally observed values: aiso(13C) ~ 47.9 MHz, A(13C) = [53.6, 45.4, 44.6] MHz; A(2HA) = [4.0, 3.3, 3.0] MHz, A(2HB) = [0.38, −0.67, −0.72] MHz. Most importantly, the carbon-bound CHO complex, model C, already ruled out by the absence of HB, is calculated to have almost three-fold and five-fold smaller 13C and 1HA hyperfine couplings, respectively: aiso (13C) = ~ 16.1 MHz, A(13C) = [23.2, 13.4, 11.7] MHz; A(2HA) = [0.51, −0.43, −0.66] MHz.
Why is the 13C coupling so large for the carbon of CH2O that is not directly coordinated to Mo(V) in the four-membered ring of model A, and not for the carbon directly bonded to the metal ion in C? We interpret this as resulting from a `transannular hyperfine interaction'.23,24 The carbon is in line with a lobe of the half-occupied Mo(dxy) orbital, and this allows overlap between Mo(dxy) and orbitals of carbon; the large aiso for 13C corresponds to only ~ 1.2% spin density in a carbon 2s orbital. Analogous behavior was first observed for a phosphorous atom that formed part of four membered cyclic structures of Mo(V) and V(IV) with dialkyl/aryldithio-phosphinato ligands.23,24 A weaker hyperfine coupling was observed when the 31P is bound to the metal-ion directly through a monodentate M-O-P linkage.25 Such considerations may have relevance to a recent proposal of a direct Fe-C bond based on a large 13C coupling.26
Finally, regarding the use of distances in the X-ray structure to infer a direct Mo-C bond, we note that the DFT-optimized cyclic structure A has a Mo-C distance of 2.76 Å, the same as found by x-ray diffraction for the glycerol-inhibited form of AOR.7
Supplementary Material
ACKNOWLEDGMENT
Support by the NIH (GM 075036, RH) and NSF (MCB0723330, BMH).
Footnotes
Supporting Information Available: One EPR figure; one ENDOR figure; DFT calculation methods.
REFERENCES
- (1).Hille R. Chemical Reviews (Washington, D. C.) 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- (2).Hille R. In Metals in Biology: Applications of High-Resolution EPR to Metalloenzymes. In: Hanson G, Berliner L, J., editors. Vol. 29. Springer; Berlin: 2010. pp. 91–121. [Google Scholar]
- (3).Pick FM, McGartoll MA, Bray RC. Eur. J. Biochem. 1971;18:65–72. doi: 10.1111/j.1432-1033.1971.tb01215.x. [DOI] [PubMed] [Google Scholar]
- (4).Howes BD, Bennett B, Bray RC, Richards RL, Lowe DJ. J. Am. Chem. Soc. 1994;116:11624–11625. [Google Scholar]
- (5).Howes BD, Bray RC, Richards RL, Turner NA, Bennett B, Lowe D. J. Biochemistry. 1996;35:1432–1443. doi: 10.1021/bi9520500. [DOI] [PubMed] [Google Scholar]
- (6).Manikandan P, Choi E-Y, Hille R, Hoffman BM. J. Am. Chem. Soc. 2001;123:2658–2663. doi: 10.1021/ja003894w. [DOI] [PubMed] [Google Scholar]
- (7).Santos-Silva T, Ferroni F, Thapper A, Marangon J, Gonzalez PJ, Rizzi AC, Moura I, Moura JJG, Romao MJ, Brondino CD. J. Am. Chem. Soc. 2009;131:7990–7998. doi: 10.1021/ja809448r. [DOI] [PubMed] [Google Scholar]
- (8).Metz S, Wang D, Thiel W. J. Am. Chem. Soc. 2009;131:4628–4640. doi: 10.1021/ja805938w. [DOI] [PubMed] [Google Scholar]
- (9).Zhang X-H, Wu Y-D. Inorg. Chem. (Washington, DC, U. S.) 2005;44:1466–1471. doi: 10.1021/ic048730l. [DOI] [PubMed] [Google Scholar]
- (10).Amano T, Ochi N, Sato H, Sakaki S. J. Am. Chem. Soc. 2007;129:8131–8138. doi: 10.1021/ja068584d. [DOI] [PubMed] [Google Scholar]
- (11).Howes BD, Pinhal NM, Turner NA, Bray RC, Anger G, Ehrenberg A, Raynor JB, Lowe D. J. Biochemistry. 1990;29:6120–6127. doi: 10.1021/bi00478a002. [DOI] [PubMed] [Google Scholar]
- (12).Morpeth FF, Bray RC. Biochemistry. 1984;23:1332–1338. doi: 10.1021/bi00301a047. [DOI] [PubMed] [Google Scholar]
- (13).Turner NA, Bray RC, Diakun GP. Biochem. J. 1989;260:563–571. doi: 10.1042/bj2600563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pauff JM, Zhang J, Bell CE, Hille R. J. Biol. Chem. 2008;283:4818–4824. doi: 10.1074/jbc.M707918200. [DOI] [PubMed] [Google Scholar]
- (15).Pauff JM, Cao H, Hille R. J. Biol. Chem. 2009;284:8760–8767. doi: 10.1074/jbc.M804517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xanthine oxidase was prepared from unpasteurized milk as described by Massey et al (1969). 2H and 13C-labeled H2CO were from Cambridge Isotopes, D2O from Isotec, Inc. 35 GHz pulsed EPR and ENDOR spectra were obtained as described by Lees et al. (2009)
- (17).Massey V, Brumby PE, Komai H, Palmer G. J. Biol. Chem. 1969;244:1682–1691. [PubMed] [Google Scholar]
- (18).Lees NS, Hänzelmann P, Hernandez HL, Subramanian S, Schindelin H, Johnson MK, Hoffman BM. J. Am. Chem. Soc. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Weber S, Kay CWM, Bacher A, Richter G, Bittl R. ChemPhysChem. 2005;6:292–299. doi: 10.1002/cphc.200400377. [DOI] [PubMed] [Google Scholar]
- (20).Kinney RA, Hetterscheid DGH, Hanna BS, Schrock RR, Hoffman BM. Inorg. Chem. (Washington, DC, U. S.) 2010;49:704–713. doi: 10.1021/ic902006v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schweiger A, Jeschke G. Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press; Oxford, UK: 2001. [Google Scholar]
- (22).Doan PE, Lees NS, Shanmugam M, Hoffman BM. Appl. Magn. Res. 2010;37:763–779. doi: 10.1007/s00723-009-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Stiefel EI, Newton WE, Pariyadath NJ. Less-Common Met. 1977;54:513–525. [Google Scholar]
- (24).Miller GA, McClung RED. Inorg. Chem. (Washington, DC, U. S.) 1973;12:2552–2561. [Google Scholar]
- (25).Gelmini L, Stephan DW. Organometallics. 1987;6:1515–1522. [Google Scholar]
- (26).Wang W, Wang K, Liu Y-L, No J-H, Li J, Nilges MJ, Oldfield E. Proc. Natl. Acad. Sci. U. S. A. 2010:1–6. doi: 10.1073/pnas.0911087107. Early Edition. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.