

Summary
In the growth plate, the interplay between Parathyroid Hormone-Related Peptide (PTHrP) and Indian Hedgehog (Ihh) signaling tightly regulates chondrocyte proliferation and differentiation during longitudinal bone growth. We found that PTHrP increases the expression of Zfp521, a zinc finger transcriptional co-regulator, in pre-hypertrophic chondrocytes. Mice with chondrocyte-targeted deletion of Zfp521 resembled PTHrP-/- and chondrocyte-specific PTHR1-/- mice, with decreased chondrocyte proliferation, early hypertrophic transition and reduced growth plate thickness. Deleting Zfp521 increased expression of Runx2 and Runx2 target genes, and decreased cyclin D1 and Bcl-2 expression while increasing caspase-3 activation and apoptosis. Zfp521 associated with Runx2 in chondrocytes, antagonizing its activity via an HDAC4-dependent mechanism. PTHrP failed to up-regulate cyclin D1 and to antagonize Runx2, Ihh and Collagen X expression when Zfp521 was absent. Thus, Zfp521 is an important PTHrP target gene that regulates growth plate chondrocyte proliferation and differentiation.
Highlights.
Zfp521 is a PTHrP target gene and effector in growth plate chondrocytes
Zfp521 antagonizes Runx2 in a HDAC4-mediated manner
Zfp521 controls chondrocyte proliferation, hypertrophy, and survival
Introduction
In the growth plate, endochondral longitudinal bone growth requires the tight regulation of entry into and exit from each stage of chondrocyte differentiation, which correspond to the well-defined resting, proliferative, pre-hypertrophic and hypertrophic zones. Alterations in this regulation disrupt the normal differentiation program, resulting in distorted growth plate architecture that leads to skeletal dysplasias with pronounced limb shortening (Kronenberg, 2003).
Parathyroid hormone-related peptide (PTHrP) is a key regulator of growth plate development (Kronenberg, 2006). It slows the progression from proliferating cells to hypertrophy by inducing cyclin D1 expression, which promotes proliferation (Beier et al., 2001; Beier et al., 1999), and by repressing expression of Runx2 (Guo et al., 2006; Iwamoto et al., 2003; Li et al., 2004), which drives chondrocyte differentiation (Kim et al., 1999; Zheng et al., 2003). In addition to slowing the transition of pre-hypertrophic cells to the hypertrophic state, PTHrP promotes the survival of hypertrophic chondrocytes by inducing the expression of the anti-apoptotic protein Bcl-2 (Amling et al., 1997). Consequently, genetic changes that eliminate (Amizuka et al., 1994; Lanske et al., 1999) or constitutively activate (Schipani et al., 1997; Weir et al., 1996) PTHrP-induced signaling result in major alterations in the structure of the growth plate and longitudinal bone growth.
Zfp521 (also known as Evi3 in mice, EHZF in humans) is a 180 kDa transcriptional co-regulator that contains 30 Krüppel-like C2H2 zinc fingers (Bond et al., 2004; Justice et al., 1994). Zfp521 is highly expressed in hematopoietic and neural stem cells. Although the role of Zfp521 in the coordination of neuronal differentiation is still elusive, it is thought to have an inhibitory function in hematopoietic stem cell differentiation, since overexpression of Zfp521 favors the expansion of hematopoietic progenitors while blocking their differentiation (Bond et al., 2004). In hematopoiesis and oncogenesis, Zfp521 exerts some of its main effects on B-cells and erythrocyte maturation by binding and antagonizing Ebf (Bond et al., 2008; Matsubara et al., 2009). In murine B-cell lymphomas, the Zfp521 gene is frequently altered by retroviral insertions adjacent to the initiation codon, which leads to increased expression of the proteins (Warming et al., 2003). Significant levels of Zfp521 transcript are also found in several human acute myeloid leukemias (Bond et al., 2008; Yamasaki et al., 2010), suggesting that altered Zfp521 activity contributes to these human hematological malignancies, possibly by increasing tumor resistance to NK cells (La Rocca et al., 2009).
We recently observed that PTHrP regulates Zfp521 expression in osteoblasts and osteocytes, where Zfp521 represses Runx2's transcriptional activity and, indirectly, expression (Wu et al., 2009). In situ hybridization also revealed Zfp521 expression around mesenchymal condensations as early as E12.5, in the perichondrium and in early chondrocytes, and in the developing growth plate (Wu et al., 2009). Based on its pattern of expression, its response to PTHrP, and its effects on Runx2, we hypothesized that Zfp521 could regulate growth plate development, possibly downstream of PTHrP. We now report that Zfp521 is an important downstream effector of PTHrP in the regulation of chondrocyte proliferation and differentiation in the growth plate, ensuring proper postnatal longitudinal bone growth.
Results
Zfp521 is highly expressed in pre-hypertrophic chondrocytes
Immunostaining of developing long bones showed expression of Zfp521 as early as E12.5 in all chondrocytes within the cartilage anlage (Figure 1A). At E14.5, Zfp521 expression decreased in the area of hypertrophic differentiation (Figure 1B). During the late stages of embryonic development (E17.5) (Figure 1C) and postnatally at 2 weeks (Figure 1H), Zfp521 was preferentially expressed within the growth plate in pre-hypertrophic chondrocytes, where it colocalized with the peak expression of the PTHrP receptor (PTHR1) (Figure 1D and 1E).
Figure 1. Zfp521 expression during skeletal development.
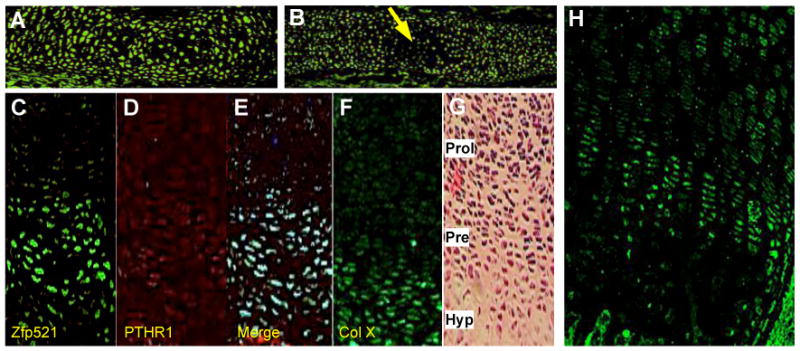
Whole tibia at E12.5 (A) (25×) and E14.5 (B) (10×) and proximal tibia growth plate at E17.5 (C-G) (10×) and 2 weeks (H) (10×) were immunostained with an antibody against Zfp521 (A,B,C,H), PTHR1 (D), or collagen type X (F) or with hematoxilin & eosin (G). (A) Zfp521 was highly expressed throughout the cartilage anlage. (B) Zfp521 staining decreased in the forming primary center of ossification (arrow). (C-E) In the formed growth plate, Zfp521 and PTHR1 were most highly expressed in pre-hypertrophic chondrocytes. Prol= proliferative, Pre= pre-hypertrophic, Hyp= hypertrophic.
Deleting Zfp521 in chondrocytes affects longitudinal bone growth and the structure of the growth plate
To examine its role in the growth plate, we deleted Zfp521 in chondrocytes using Cre recombinase driven by the collagen II promoter (Col II-Cre) (Long et al., 2001). The deletion efficiency was confirmed in primary rib chondrocytes by qPCR (Figure S1A). Although Zfp521fl/fl Cre+ (cKO) mice were similar to floxed-control (Zfp521fl/fl Cre-) mice at birth (Figure S1B), growth retardation was evident by 1-2 weeks of age (Figure 2A) and still present at 8 weeks (Figure S1E) and 12 weeks (not shown). Consistent with the pattern of Col II expression, bones that formed by endochondral ossification were smaller while craniofacial structures that develop by intramembranous ossification and the cranium base synchondroses appeared normal (Figure S1C and S1D). No other gross anatomical alterations were seen in the Zfp521 cKO mice.
Figure 2. Deleting Zfp521 in chondrocytes alters growth plate structure and skeletal development.
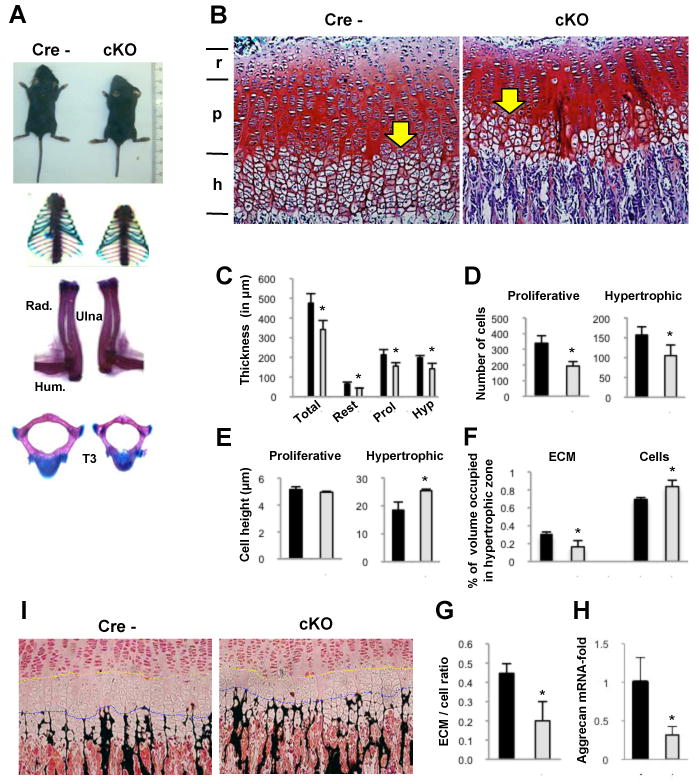
(A) Two-week-old Zfp521 cKO mice (right) exhibited postnatal dwarfism. Rib cage, long bones and vertebrae of the Zfp521 cKO mice were smaller than in the floxed control.
(B) Proximal tibia growth plates from 2 week-old mice were stained with Safranin O (10×). A reduction in the thickness of all layers was observed in the Zfp521 cKO growth plates (r, resting; p, proliferative; h, hypertrophic). In addition, the hypertrophic zone was closer to the epiphysis (yellow arrow) suggesting early hypertrophic differentiation.
(C-H) Data derived from Zfp521 cKO mice are shown in white and data obtained in floxed control mice are presented in black (C) Histomorphometric analysis of proximal tibia growth plates from the Zfp521 cKO and floxed control mice confirmed an approximate 30% reduction in the thickness of all layers. Also, there were fewer cell columns with fewer cells per column in the proliferative zone (339±48 vs 193±28) and a reduced cell population in the hypertrophic zone (157±21 vs 105±27) (D). (E) The cell height of hypertrophic chondrocytes in Zfp521 cKO growth plates was increased (17.7±2.7μm vs 23.7±2.9μm), whereas proliferative chondrocytes were similar in floxed control and mutant growth plates. (F) The percentage of the hypertrophic zone volume occupied by extracellular matrix (ECM) was reduced in mutant growth plates (31±0.02% vs 16±0.07%) a comparable increase in the percentage occupied by cells (69±0.02% vs 84±0.07%), which generated a decrease in the ECM/cell ratio (0.44±0.05 vs 0.20±0.07) (G). (H) The expression of aggrecan mRNA is reduced by about 50% in the growth plate of Zfp521 cKO mice.
(I) Von Kossa staining showed earlier chondroid mineralization (black) in the Zfp521 cKO growth plates. Note the reduction in the number of layers of hypertrophic chondrocytes between the pre-hypertrophic chondrocytes and the leading edge of the mineralization front (yellow and blue lines). Data are means ± S.D., n=4, P<0.05.
Histomorphometric analysis of proximal tibia from 2 week-old mice revealed a 30% reduction in the mutant growth plate thickness, affecting all zones (Figure 2B and 2C). The resting zone contained only one or two layers of cells, rather than five or more layers in the Zfp521fl/flCre- growth plates. Proliferating cells were properly oriented along the long axis of the bone and had a normal flat morphology, but the numbers of cell columns and cells per column were reduced (Figure 2D). The transition to the hypertrophic zone was closer to the epiphysis. The hypertrophic zone thickness was also reduced (Figure 2B and 2C) with fewer chondrocytes, each of increased cellular height (Figure 2D and 2E), and about 50% less extracellular matrix (ECM) (Figure 2F and 2H), resulting in a reduced matrix-to-cell ratio (Figure 2G), suggesting impaired matrix production. The onset of ECM mineralization also occurred prematurely (Figure 2I), due to decreased proliferation, accelerated differentiation or both. Consequently, while an average of six layers of hypertrophic chondrocytes were present between the pre-hypertrophic chondrocytes and the leading edge of the mineralization front in the Zfp521fl/fl Cre-, only three layers were present in the cKO mice.
In situ hybridization (ISH) confirmed the altered growth plate architecture in the absence of Zfp521 (Figure 3). Reduction in the thickness of the entire growth plate and the proliferative and hypertrophic zones were revealed by Col II expression (Figure 3A), the hedgehog receptor subunit patched (Ptc) (Figure 3B) and Col X, respectively (Figure 3C). Ihh and PTHR1, both markers of pre-hypertrophic cells, were upregulated and located closer to the epiphysis than in the control (Figure 3D and 3E). Quantitative PCR of mRNA isolated from Zfp521 cKO and Zfp521fl/fl Cre- mice growth plate chondrocytes confirmed the ISH results (Figure 3F-3J).
Figure 3. In situ hybridization of differentiation markers.
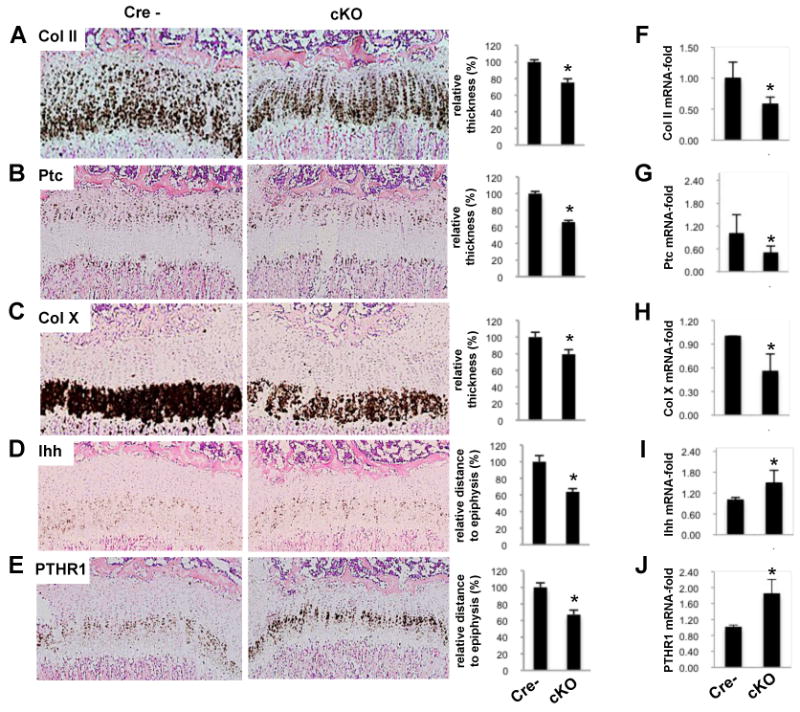
(A-E) In situ hybridization (left panels) including quantification (right panels) of proximal tibiae from 2 week-old Zfp521 cKO and floxed control (Cre-) mice (10×). (A) The collagen type II (Col II) signal in the cKO growth plate was thinner (25±2% reduction), demonstrating the overall thinning of the growth plate. The signals for patched (Ptc) (B) and collagen type × (Col X) (C) revealed the narrower proliferative (34.3±3% reduction) and hypertrophic (20.7±3% reduction) zones respectively. Pre-hypertrophy markers Ihh (D) and PTHR1 (E) demonstrated the closer proximity of the pre-hypertrophic zone to the epiphysis (35.9±2% and 32.9±3% reduced distance between markers and top of the growth plate respectively). Quantification of the granular area fraction covered by Col X, PTHR1 and Ihh signal, as described in Experimental Procedures, revealed changes of 6-fold reduction, 3-fold increase and 1.7-fold increase, respectively, in the Zfp521 cKO growth plate.
(F-J) qPCR of mRNA isolated from floxed and mutant growth plate chondrocytes are presented.
Zfp521 regulates chondrocyte proliferation
Cell proliferation was decreased in the Zfp521 cKO growth plate (40±5% BrdU-positive cells in Zfp521fl/fl Cre- mice, 28±3% in the cKO mice, p<0.005) (Figure 4A and 4B). In ATDC5 cells, depleting Zfp521 with shRNA (Figure S2A) increased the doubling time and decreased both micromass size and BrdU incorporation (Figure S2B-S2D), while forced expression of Zfp521 increased BrdU incorporation by about 50% (Figure S2E and S2F). Neither transient overexpression of Zfp521 in fibroblastic NIH3T3 cells nor stable depletion of Zfp521 in osteoblastic MC3T3-E1 cells altered the BrdU incorporation (Figure S2G and S2H), demonstrating that the effect of Zfp521 on cell proliferation is chondrocyte-specific.
Figure 4. Zfp521 regulates chondrocyte proliferation and the expression of hypertrophy and terminal differentiation markers.
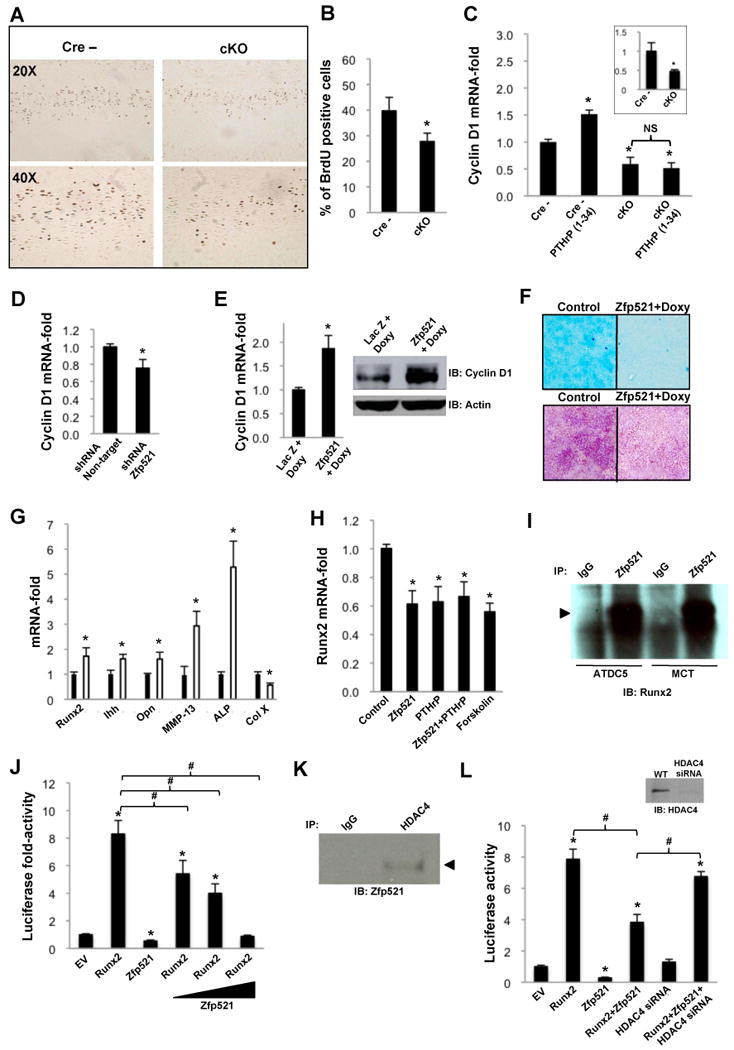
(A) Two-week-old floxed control and Zfp521 cKO mice were injected with BrdU labeling solution (1ml/100g body weight) 20 and 4 hours before sacrifice and proximal tibia were immunostained for BrdU.
(B) Zfp521 cKO growth plates contained significantly fewer BrdU-positive chondrocytes than controls.
(C) Rib chondrocytes and growth plate chondrocytes (insert) isolated from floxed control and cKO mice were cultured with or without PTHrP (1-34) (100 nM) for 48h. The lack of Zfp521 decreased cyclin D1 expression in untreated chondrocytes and abolished the PTHrP (1-34)-induced increase in cyclin D1 expression.
(D) Zfp521 shRNA (seq.4) reduced cyclin D1 expression in ATDC5 cells cultured in micromass for 10 days.
(E) Doxycycline-induced overexpression of Zfp521 in micromass cultures of ATDC5 cells stably transfected with Tet-inducible Zfp521 increased cyclin D1 mRNA (graph) and protein (Western blot).
(F) Tet-inducible Zfp521-overexpressing ATDC5 cells and LacZ control cells were cultured in the presence of 0.5 μg/ml doxycycline for 16 d and stained with Alcian blue (upper panel) or from d16 to d30 and stained for ALP activity (lower panel). Overexpressing Zfp521 repressed both the formation of Alcian blue-positive cartilage nodules and ALP activity.
(G) Primary rib chondrocytes isolated from floxed controls (black bars) and Zfp521 cKO mice (white bars) were cultured for 3 days in maintenance medium and the expression of Runx2 and its target genes was analyzed by qPCR. The expression of Runx2 and all target genes except Col X were increased in the Zfp521 cKO chondrocytes. Col X expression was reduced by ∼50%.
(H) Overexpressing Zfp521 in MCT cells reduced Runx2 expression to the same extent as PTHrP (1-34) (100 nM) and forskolin (10 μM), and the effects were not additive.
I) Co-immunoprecipitation of endogenous Zfp521 and Runx2 in ATDC5 and MCT cells.
J) 6xOSE2-luc reporter assay showing Zfp521 dose-dependently repressing the inductive activity of Runx2 in MCT cells.
(K) Co-immunoprecipitation of endogenous Zfp521 and HDAC4 in ATDC5 cells.
(L) 6xOSE2-luc reporter assay demonstrating that Zfp521 requires HDAC4 to effectively repress Runx2 transcriptional activity in MCT cells. Inset, HDAC4 knock-down efficiency by siRNA transiently transfected into MCT cells before hypertrophic induction. (B,C,D,E,G,H,J,L) Data are means ± S.D., n=3, *p<0.05, significant difference from control, #p<0.05 significant difference from indicated condition.
Cyclin D1 promotes chondrocyte proliferation and cyclin D1-deficient mice exhibit a thin proliferative zone (Beier et al., 2001). In primary chondrocytes from Zfp521 cKO mice, we found decreased proliferation (data not shown), confirming our in vivo results, and decreased expression of cyclin D1 (Figure 4C). Similarly, depleting Zfp521 reduced cyclin D1 expression in ATDC5 cells (Figure 4D) while forced expression of Zfp521 increased both mRNA and protein levels of cyclin D1 (Figure 4E). These results suggest that Zfp521 promotes cyclin D1 expression, contributing to keeping growth plate chondrocytes in a proliferative state, although we found that Zfp521 did not affect the activity of a cyclin D1-luciferase reporter gene in ATDC5 cells (data not shown), suggesting that the changes in cyclin D1 expression that occur when Zfp521 is depleted or overexpressed is not the result of a direct effect on the cyclin D1 promoter.
Zfp521 slows the progression of chondrocytes to hypertrophy
Since growth plate development requires a balance between chondrocyte proliferation and progression to hypertrophy, we next examined the effect of Zfp521 on alkaline phosphatase (ALP) activity, an early hypertrophy marker. Overexpressing a tetracycline (tet)-inducible Zfp521 in ATDC5 cells from day 0 until day 16 of culture, i.e. before the transition to hypertrophy, repressed the formation of Alcian blue-positive cartilage nodules (Figure 4F upper panel), suggesting impaired chondrogenic differentiation. Overexpression from day 16 until day 30, i.e. at the time of hypertrophic transition and beyond, induced a significant decrease in ALP activity (Figure 4F lower panel). Thus, Zfp521 appears to antagonize hypertrophic differentiation as well as promoting chondrocyte proliferation.
Zfp521 antagonizes Runx2 activity in chondrocytes
Runx2 is a key promoter of chondrocyte hypertrophy. We therefore assayed the expression of Runx2 and its target genes in cultured primary rib chondrocytes from Zfp521 cKO and Zfp521fl/fl Cre- mice. The expression of Runx2, Ihh, osteopontin (OPN), ALP and MMP-13 all increased in the absence of Zfp521 (Figure 4G). Consistent with the ISH results, and despite the fact that it is also considered a Runx2 target gene, expression of the matrix component Col X was decreased by about 50%, suggesting that Zfp521 also regulates Col X gene expression by Runx2-independent mechanisms.
We next examined the effect of Zfp521 on Runx2 expression in MCT cells, an immortalized chondrocyte cell line that rapidly becomes hypertrophic at non-permissive temperatures (Zheng et al., 2003). As expected, inducing hypertrophic differentiation increased the expression of Runx2 significantly (not shown). Transiently transfecting MCT cells with Zfp521 prior to inducing hypertrophy reduced Runx2 as much as treating with PTHrP and Forskolin (Figure 4H), confirming the ability of Zfp521 to suppress Runx2 expression.
We have previously shown that Zfp521 associates with Runx2 and antagonizes its transcriptional activity in osteoblasts (Wu et al., 2009). Endogenous Zfp521 also immunoprecipitated with Runx2 from ATDC5 and MCT cells (Figure 4I). We therefore examined whether Zfp521 also regulated Runx2 activity in hypertrophic chondrocytes, and found that Zfp521 dose-dependently repressed Runx2-induced activation of the 6xOSE2-luc reporter plasmid in MCT cells during temperature-induced hypertrophic differentiation, (Figure 4J), indicating that Zfp521 also controls Runx2 transcriptional activity in differentiating chondrocytes.
Repression of chondrocyte Runx2 activity by Zfp521 requires HDAC4
Several histone deacetylases (HDACs) antagonize Runx2 (Kang et al., 2005; Schroeder et al., 2004; Westendorf et al., 2002) and it has been reported that HDAC4 binds and antagonizes Runx2 in pre-hypertrophic chondrocytes (Vega et al., 2004) and that PTHrP induces the nuclear translocation of HDAC4 (Kozhemyakina et al., 2009). We therefore looked for a possible connection between Zfp521 and HDAC4, and found that the two proteins form an endogenous complex in ATDC5 cells (Figure 4K) and that depleting HDAC4 blunted the Zfp521-induced Runx2 repression (Figure 4L).
Zfp521 regulates chondrocyte apoptosis
Apoptosis of hypertrophic chondrocytes is another key event in growth plate development. The reduced number of hypertrophic cells suggested that Zfp521 might antagonize chondrocyte apoptosis. As hypothesized, the number of apoptotic hypertrophic chondrocytes increased by 2-fold in the Zfp521 cKO growth plate (Figure 5A). Induction of apoptosis culminates in increased caspase activity, especially caspase-3 (Porter and Janicke, 1999), and indeed, caspase activity was increased in Zfp521 cKO primary chondrocytes (Figure 5B top panels). Similarly, caspase activity and activated caspase-3 protein in ATDC5 cells were increased in by depleting Zfp521 (Figure 5B middle panels and Figure 5C) and decreased by overexpressing Zfp521 (Figure 5B bottom panels and Figure 5D). Finally, we found that expression of the anti-apoptotic protein Bcl-2, which enhances survival of terminally differentiated chondrocytes (Amling et al., 1997), was significantly decreased in primary chondrocytes from Zfp521 cKO mice (Figure 5E). Thus, our results suggest that Zfp521 antagonizes chondrocyte apoptosis.
Figure 5. Zfp521 regulates chondrocyte apoptosis.
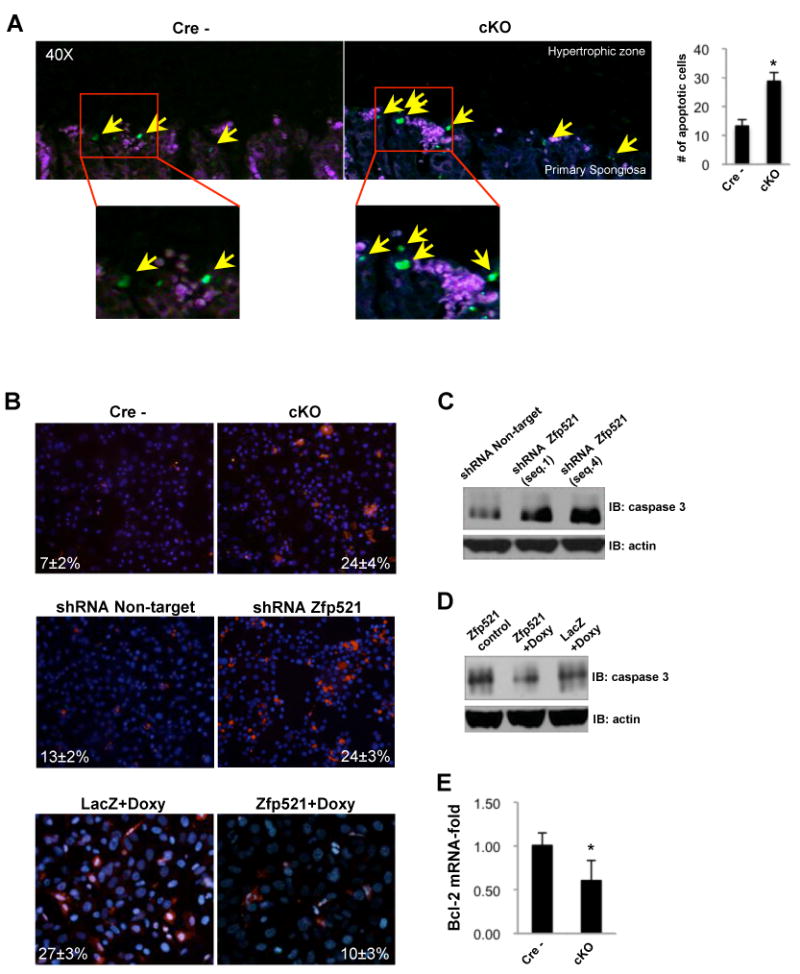
(A) TUNEL assay in 3 week-old proximal tibias from Cre- and cKO mice demonstrating an in vivo increased (∼3-fold) chondrocyte apoptosis in the absence of Zfp521. Apoptotic cells are indicated (arrows).
(B) Primary rib chondrocytes isolated from Zfp521 cKO mice (top panels), Zfp521 shRNA-expressing ATDC5 cells (seq.4) (middle panels) and Tet-inducible Zfp52-overexpressing ATDC5 cells (bottom panels) and respective controls were stained for active caspase with FLICA (red). Nuclei were counterstained with Hoechst stain (blue). Caspase activity was increased by deleting/depleting Zfp521 and decreased by overexpressing Zfp521. The percentage of caspase-positive cells is indicated.
(C) Western blot of total cell lysates showed increased activated caspase-3 in Zfp521 shRNA-expressing ATDC5 cells (2 different sequences) cultured for 12 days in chondrogenic conditions.
(D) Activated caspase-3 was decreased in doxycycline (0.5 μg/ml)-treated Tet-inducible Zfp521-overexpressing ATDC5 cells.
(E) Bcl-2 mRNA expression was decreased in primary rib chondrocytes isolated from Zfp521 cKO mice. Data are means ± S.D., n=3, *p<0.05, significant difference from floxed control.
PTHrP regulates Zfp521 expression in differentiating chondrocytes
Our results consistently demonstrated that Zfp521 regulates growth plate chondrocyte proliferation, differentiation and apoptosis. The reduced proliferation and the early transition to hypertrophy in Zfp521 cKO growth plates are reminiscent of the effects of decreased PTHrP signaling in chondrocytes (Amizuka et al., 1994; Kobayashi et al., 2002; Lanske et al., 1999). We therefore examined whether Zfp521 played a role in the responses of chondrocytes to PTHrP.
Consistent with the in vivo localization of Zfp521 in pre-hypertrophic chondrocytes (Figure 1H), the expression of endogenous Zfp521 paralleled the temporal and spatial expression of endogenous PTHR1 in ATDC5 micromass and pellet cultures (Figure S3), with peaks of expression coinciding during the hypertrophic transition of ATDC5 cells and the highest responsiveness to PTHrP (Shukunami et al., 1996). Transient over-expression of Zfp521 in ATDC5 micromass cultures caused a significant decrease in the number of Alcian blue-positive nodules after 2 weeks, comparable to the decrease in cultures treated with PTHrP (1-34) (Figure 6A). The inhibitory effects of PTHrP (1-34) and Zfp521 on nodule formation were not additive, suggesting that PTHrP and Zfp521 share the same signaling pathway.
Figure 6. PTHrP regulates Zfp521 expression in differentiating chondrocytes.
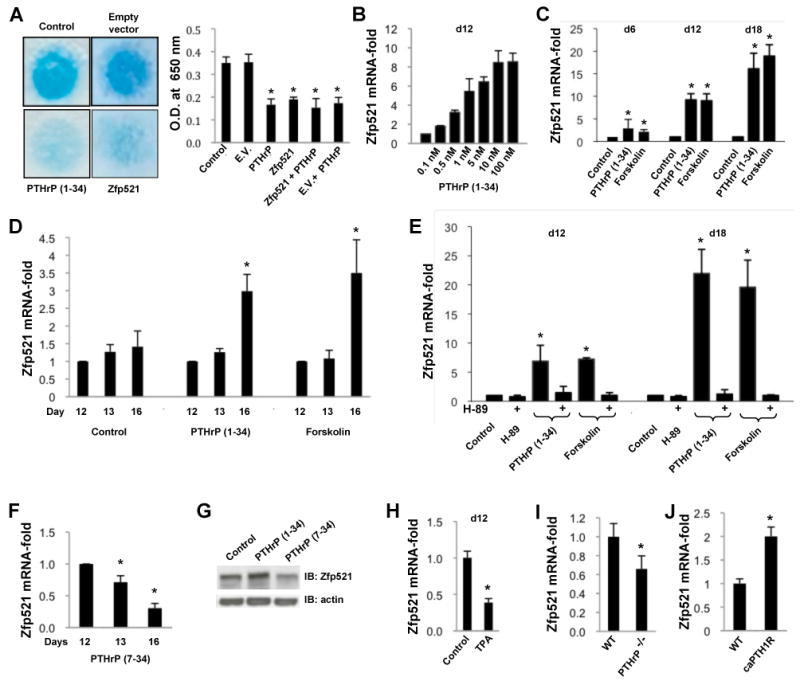
(A) Transiently transfected Zfp521 reduced the formation of Alcian blue-stained cartilage nodules in 12 day ATDC5 cell micromass cultures to the same extent as 100 nM PTHrP (1-34) (E.V. = empty vector). Alcian blue staining was quantified spectrophotometrically after guanidine extraction.
(B) Continuous treatment of ATDC5 cells with PTHrP for 12 days dose-dependently increased Zfp521 mRNA expression.
(C, D) PTHrP (1-34) (100 nM) and forskolin (10 μM) increased Zfp521 mRNA expression in a time-dependent manner in ATDC5 cells treated either continuously for 6, 12 or 18 days (C) or at the beginning of hypertrophic conversion (d12 to d16) (D).
(E) The PKA inhibitor H-89 (10 μM) prevented the PTHrP- and forskolin-induced increase in Zfp521 mRNA expression in ATDC5 cells.
(F, G) Treatment of ATDC5 cells with 100 nM PTHrP (7-34) at the beginning of hypertrophic transition downregulated Zfp521 mRNA (F) and protein (G) levels at d16.
(H) Treating ATDC5 cells with TPA (1 μM) continuously for 12 days to activate PKC downregulated the expression of Zfp521 mRNA.
(I, J) Zfp521 mRNA was decreased in primary rib chondrocytes isolated from PTHrP-/- mice (I) and increased in primary rib chondrocytes from mice that overexpress constitutively active PTHR1 (caPTHR1) (J), confirming that PTHrP/PTHR1 signaling induces Zfp521 expression. Data are means ± S.D., n=5, *p<0.05, significant difference from control.
Given the similar expression patterns of Zfp521 and PTHR1 in chondrocytes in vivo and in vitro, and their comparable inhibitory effects on chondrocyte differentiation, we hypothesized that Zfp521 could be an effector downstream of PTHrP. Indeed, PTHrP (1-34) increased Zfp521 expression dose-dependently (Figure 6B) and time-dependently (Figure 6C and 6D). Forskolin had a similar effect, and the selective PKA inhibitor H-89 prevented the induction of Zfp521 expression by both PTHrP (1-34) and forskolin (Figure 6E), indicating that the Gαs/cAMP/PKA signaling pathway couples PTHrP to Zfp521 expression.
In contrast to the effect of PTHrP (1-34), PTHrP (7-34), which selectively activates PLC-γ signaling (Nutt et al., 1990), down-regulated Zfp521 mRNA and protein levels in a similar time-dependent manner (Figure 6F and 6G). Stimulating protein kinase C directly with 12-O-tetradecanoylphorbol-13-acetate (TPA) achieved a similar reduction (Figure 6H), suggesting that the PTHrP-induced PLC-γ signaling pathway might oppose cAMP/PKA in the regulation of Zfp521 downstream of PTHR1.
Zfp521 expression was also reduced in primary rib chondrocytes from PTHrP-/- mice (Figure 6I) and increased in primary rib chondrocytes from mice that overexpress the mutated human PTHR1 that constitutively activates adenylyl cyclase (Schipani et al., 1997) (Figure 6J). Together with the results from the ATDC5 cultures, these data firmly establish that PTHrP induces the expression of Zfp521 in chondrocytes.
Zfp521 contributes to the effects of PTHrP on chondrocytes
PTHrP induces cyclin D1 expression and proliferation (Beier et al., 2001; Beier et al., 1999) and down-regulates Runx2 expression (Guo et al., 2006; Li et al., 2004) (Figure 7A), while deleting Zfp521 decreased proliferation and cyclin D1 expression and increased Runx2 expression. The induction of Zfp521 expression by PTHrP suggested that Zfp521 mediated at least part of PTHrP's effects on chondrocyte proliferation and Runx2 expression. As hypothesized, we found that the loss of Zfp521 prevented the PTHrP (1-34)-induced up-regulation of cyclin D1 mRNA (Figure 4C). We also found that although PTHrP (1-34) down-regulated the expression of Runx2 in ATDC5 cells at the beginning of the hypertrophic conversion (Figure 7A) and in primary chondrocytes from Zfp521fl/fl Cre- mice (Figure 7B) as expected, it failed to reduce Runx2 expression when Zfp521 was absent (Figure 7B and 7C). Finally, the absence of Zfp521 also increased constitutive Ihh expression in primary chondrocytes and prevented PTHrP's down-regulation of Ihh (Figure 7D), identifying another downstream response to PTHrP that requires Zfp521 and indicating the potential importance of Zfp521 in the negative feedback loop that controls hypertrophy.
Figure 7. Zfp521 mediates some effects of PTHrP on chondrocytes.
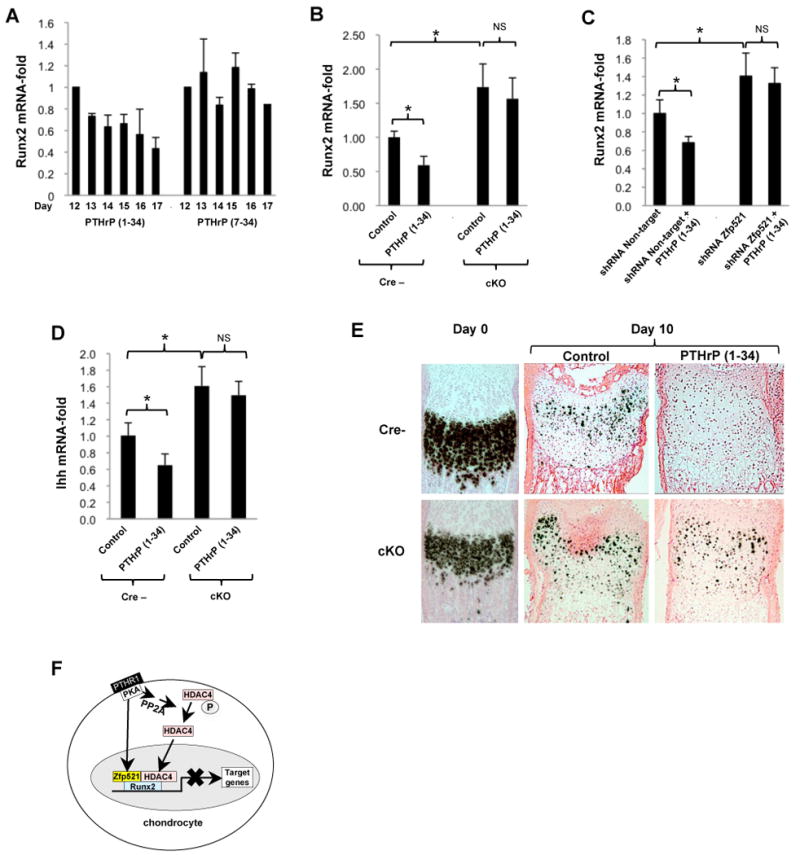
(A) Micromass cultures of ATDC5 cells were exposed to 100 nM PTHrP (1-34) or PTHrP (7-34) at the onset of hypertrophic conversion (d12 to d16). PTHrP (1-34) induced a progressive decrease in Runx2 mRNA expression, but PTHrP (7-34) had no effect.
(B) Primary rib chondrocytes were isolated from 2-week-old Zfp521 cKO and floxed control mice and treated for 48h with 100 nM PTHrP (1-34). PTHrP failed to repress Runx2 mRNA expression in the Zfp521 cKO cells (NS = not significant).
(C) Micromass cultures of ATDC5 cells stably expressing Zfp521 (seq.4) or non-target shRNAs were treated with 100 nM PTHrP (1-34) for 48h. Depleting Zfp521 prevented the PTHrP-induced decrease in Runx2 expression.
(D) Ihh expression was measured in primary chondrocytes from 2-week-old Zfp521 cKO and floxed control mice treated for 48h with 100 nM PTHrP (1-34). PTHrP failed to repress Ihh mRNA expression in the Zfp521 cKO cells.
(E) Metatarsals from 2 weeks-old Zfp521 cKO and floxed control mice were cultured with or without PTHrP (1-34) for 10 days and assayed for Col X mRNA by in situ hybridization. The absence of Zfp521 blunted the repressive effect of PTHrP on Col X expression. Note the presence of the growth plate phenotype on mutant metatarsals both at day 0 and day 10, and the alterations of the growth plate morphology of both wild-type and mutant mice at day 10 due to in vitro assay conditions.
(F) Proposed model of action of Zfp521 in pre-hypertrophic chondrocytes. In chondrocytes, activation of the parathyroid hormone (PTH) receptor 1 (PTHR1) causes a PKA-mediated increase in Zfp521 expression and activates protein phosphatase 2A, which dephosphorylates HDAC4 (Kozhemyakina et al., 2009), causing its nuclear accumulation. In the nucleus, Zfp521 forms a complex with HDAC4 and Runx2, leading to a repression of Runx2-mediated target gene activation. (A-D) Data are means ± S.D., n=3, *p<0.05.
To evaluate the effect of Zfp521 deletion on PTHrP's control of hypertrophy, metatarsals from 2-week-old Zfp521 cKO and control Zfp521fl/fl Cre- mice were cultured for 10 days with or without PTHrP (1-34), and expression of Col X, a hypertrophic marker that is down-regulated by PTHrP, was examined by ISH (Figure 7E). PTHrP effectively repressed the expression of Col X mRNA in controls, but deletion of Zfp521 blunted the effect of PTHrP on Col X expression, providing more evidence of the role of Zfp521 downstream of PTHrP in controlling the transition to hypertrophy in the growth plate.
Zfp521 represses PTHR1 expression in pre-hypertrophic chondrocytes
In addition to Zfp521 expression being regulated by PTHrP downstream of PTHR1, we observed that PTHR1 expression was markedly increased in the Zfp521 cKO growth plate (Figure 3E and 3J), suggesting that Zfp521 down-regulates PTHR1 expression in pre-hypertrophic chondrocytes. We confirmed this by showing that depleting Zfp521 in ATDC5 cells increased PTHR1 protein (Figure S4A), while overexpressing Zfp521 in ATDC5 cells and MCT cells reduced PTHR1 protein (Figures S4B, S4C). These data suggest that Zfp521 mediates a negative feedback loop that down-regulates PTHR1 expression in pre-hypertrophic chondrocytes, thereby further contributing to the regulation of PTHrP effects on the growth plate.
Discussion
Postnatal longitudinal bone growth depends on chondrocyte proliferation and differentiation in the growth plate, a process that is tightly controlled by PTHrP and Ihh (Kronenberg, 2003). Zfp521 is a 180 kDa 30 Krüppel-like zinc finger protein that regulates the differentiation of neurons, hematopoietic cells and osteoblasts (Bond et al., 2004; Warming et al., 2003; Wu et al., 2009). We now report that Zfp521 is also expressed in growth plate chondrocytes and plays a role in the regulation of their proliferation, differentiation and apoptosis in response to PTHrP. Targeted deletion of chondrocyte Zfp521 led to postnatal growth retardation with reduced thickness of all growth plate zones. Zfp521 cKO chondrocytes exhibited decreased proliferation, early hypertrophic transition, reduced matrix production and enhanced apoptosis. Deletion of Zfp521 also decreased the expression of cyclin D1 and increased the expression of Runx2 and several Runx2 target genes. Also, Bcl-2 expression decreased and caspase-3 activation increased when Zfp521 was absent. The decreased proliferation and consequent reduction in the number of cells entering the differentiation program is a likely key contributor to the reduced thickness of all the zones of the Zfp521 cKO growth plate. In addition, the reduced matrix production and increased apoptosis would also contribute to the thinning of the growth plate.
PTHrP is an essential regulator of chondrocyte proliferation, differentiation and life span in the growth plate during longitudinal bone growth (Kobayashi et al., 2002; Kronenberg, 2006). It controls the expression of at least three key regulatory proteins: cyclin D1 is increased to maintain proliferation of early stage chondrocytes (Beier et al., 2001; Beier et al., 1999); Runx2 (and secondarily Ihh) expression is decreased to delay the onset of the hypertrophic transition and initiation of matrix mineralization (Guo et al., 2006), and Bcl-2 is increased to delay apoptosis of hypertrophic chondrocytes (Amling et al., 1997).
Our results indicate that PTHrP also regulates the expression of Zfp521 and that Zfp521 mediates some of the important downstream effects of PTHrP in the growth plate. First, several effects of overexpressing Zfp521 in chondrocytes resemble changes induced by PTHrP, while the Zfp521 cKO growth plate resembles the growth plates of the PTHrP KO and chondrocyte-specific PTHR1 KO mice (Amizuka et al., 1994; Kobayashi et al., 2002). Second, PTHrP induced the expression of Zfp521 in chondrocytes in a dose- and time-dependent manner, similar to our observation in osteoblasts (Matsubara et al., 2009; Wu et al., 2009). Zfp521 expression in primary chondrocytes was markedly decreased by deleting PTHrP and increased by expressing the mutated PTHR1 that constitutively activates adenylyl cyclase. Third, PTHrP failed to induce cyclin D1 expression and to repress Runx2, Ihh, and Col X expression in chondrocytes when Zfp521 was absent, and Bcl2 expression, which is induced by PTHrP in chondrocytes (Amling et al., 1997), was also reduced in the absence of Zfp521. Thus, Zfp521 is a PTHrP target gene that appears to be a key effector of PTHrP in mechanisms that modulate chondrocyte proliferation, differentiation and apoptosis in the growth plate.
The decreased cell proliferation is chondrocyte-specific as it was not seen in fibroblastic- or osteoblastic cells and it is consistent with reports that Zfp521 acts to prevent differentiation and increase early precursor pools within the hematopoietic lineage (Bond et al., 2004; Warming et al., 2003). Zfp521 may therefore function as a repressor of exit from the cell cycle and progression to more mature cells. Zfp521 may contribute to the regulation of chondrocyte proliferation by at least two mechanisms. First, Zfp521 promotes basal cyclin D1 expression in primary chondrocytes, which is required for normal proliferation of growth plate chondrocytes, and mediates the PTHrP-induced increase in cyclin D1 expression, although we found that Zfp521 did not affect the activity of a cyclin D1-luciferase reporter gene in ATDC5 cells (data not shown), suggesting that the changes in cyclin D1 expression that occur when Zfp521 is depleted or overexpressed is not the result of a direct effect on the cyclin D1 promoter. The mechanism could involve modulating the coupling of PTHrP to CREB (Beier et al., 2001) or possibly AP-1 factors, which we find bind to Zfp521 (unpublished observation), or other factors (e.g., Ebf1) that have been reported to interact with Zfp521. However, cyclin D1 expression and chondrocyte proliferation are also induced by Ihh (Long et al., 2001) and we cannot rule out a role for Zfp521 downstream of Ihh. Second, the inability of PTHrP to antagonize Runx2 expression in the absence of Zfp521 and the resulting elevated Runx2 expression could also contribute to the reduced size of the proliferative zone, since overexpressing Runx2 in proliferating and prehypertrophic chondrocytes reduces proliferation and the thickness of the proliferative zone (Takeda et al., 2001).
The repression of Runx2 expression by PTHrP, mediated by cAMP/PKA, is a critical event in the normal regulation of the chondrocyte hypertrophic transition (Iwamoto et al., 2003; Li et al., 2004). Mice that lack Runx2 or overexpress a dominant-negative form of Runx2 exhibit no hypertrophic conversion (Inada et al., 1999; Ueta et al., 2001), while targeted overexpression of Runx2 in pre-hypertrophic chondrocytes accelerates hypertrophic differentiation (Takeda et al., 2001). We confirmed that PTHrP-induced activation of the cAMP/PKA pathway in chondrocytes represses Runx2 expression, and showed that it also increases Zfp521 expression. More importantly, we found that PTHrP failed to repress Runx2 expression in chondrocytes when Zfp521 was absent. Consistent with the inhibitory effect of Zfp521 on Runx2 transcriptional activity (Wu et al., 2009), hypertrophic conversion was accelerated in the Zfp521 cKO mice, with increased expression of ALP, Runx2 and the Runx2 target genes Ihh, MMP-13 and OPN, all markers of chondrocyte differentiation, strongly suggesting that Zfp521 contributes to the PTHrP-induced repression of Runx2 expression and activity in chondrocytes, and the resulting delay of hypertrophy.
HDACs inhibit gene expression by deacetylating both histones (class I HDACs) and transcription factors (class II HDACs) (Haberland et al., 2009). We found that the repression of Runx2 activity by Zfp521 requires HDAC4. This class II HDAC represses the transcriptional activity of Runx2 and MEF2C, another key regulatory factor in pre-hypertrophic chondrocytes, and HDAC4-null mice display premature chondrocyte hypertrophy (Vega et al., 2004), much like the effect of deleting Zfp521 (this study) or the constitutive expression of Runx2 or MEF2C in chondrocytes (Arnold et al., 2007). PTHrP induces the nuclear translocation and repression of gene expression by HDAC4 (Kozhemyakina et al., 2009). Our findings (1) that PTHrP also induces Zfp521 expression and (2) that Zfp521 inhibits Runx2 transcriptional activity in an HDAC4-dependent manner add another key element to this important growth plate regulatory mechanism.
The last steps in the progression of chondrocyte differentiation in the growth plate are the apoptosis of hypertrophic cells and the replacement of cartilage by bone. PTHrP slows the onset of chondrocyte apoptosis (Amizuka et al., 2004; Amizuka et al., 1996; Yamanaka et al., 2003), consistent with our report that PTHrP signaling increases the expression of the anti-apoptotic molecule Bcl-2 (Amling et al., 1997). Conversely, mice with Col II promoter-driven Runx2 overexpression exhibit accelerated hypertrophic differentiation and premature cartilage mineralization (Ueta et al. 2001), and overexpression of Runx2 in primary chondrocytes increased matrix calcification (Enomoto et al., 2000). Consistent with the increased Runx2, we found that apoptosis of terminally differentiated chondrocytes was increased in the Zfp521 cKO growth plates, while Bcl-2 expression was decreased and caspase-3 activation was increased in Zfp521 cKO primary chondrocytes. This increased cell death presumably contributes to the thinner hypertrophic zone of the Zfp521 cKO growth plate, together with the reduced proliferation and production of matrix proteins and the accelerated hypertrophic conversion.
Although the growth plate phenotype of the Zfp521 cKO mice resembles that of the Col II-Cre: PTHR1fl/fl mice, it is less severe, suggesting that not all PTHrP-induced regulatory mechanisms depend on Zfp521 or that Zfp521 also modulates PTHrP-independent mechanisms that regulate growth plate development. A major difference is the time of presentation. Zfp521 cKO newborns were little different from control littermates, in contrast to the Col II-Cre: PTHR1fl/fl mice in which the shortening of the growth plate was well established by E17.5 (Kobayashi et al., 2002). The lack of an embryonic phenotype in chondrocyte-specific Zfp521 deletion was unexpected, given the clear co-expression of Zfp521 and PTHR1 in prehypertrophic cells as early at E17.5. However, deregulation or deletion of other regulatory molecules, e.g., constitutively active FGFR3 mutants (Chen et al., 1999) and deletion of the growth hormone (GH) receptor (Lupu et al., 2001), led to phenotypes that appear only postnatally. Most relevant for this study, deleting cyclin D1 (Beier et al., 2001) and overexpressing Runx2 (Takeda et al., 2001) both cause a postnatal thinning of the growth plate similar to that in the Zfp521 cKO mice, consistent with functional interaction of the molecules. The absence of a phenotype seen in the Runx2-overexpressing mice in late embryonic and perinatal stages, even though endogenous Runx2 expression is highest at E14.5 (Inada et al., 1999; Takeda et al., 2001), suggests that different Runx2-dependent mechanisms regulate growth plate chondrocyte development in embryonic and postnatal growth plates, and that growth plate chondrocyte differentiation may be more sensitive to high levels of Runx2 postnatally. Compensation during embryogenesis for the loss of Zfp521 by the related Zfp423, which is also expressed in chondrocytes, could also contribute to the late onset of the phenotype.
In conclusion, our results demonstrate that the 30 Krüppel-like zinc finger protein Zfp521 contributes significantly to the regulation of proliferation, hypertrophy and apoptosis of growth plate chondrocytes, thereby playing an important role in longitudinal bone growth. Zfp521 accomplishes this in part by mediating PTHrP-induced inhibition of Runx2 expression in prehypertrophic chondrocytes in an HDAC4-dependent mechanism (Figure 7F). Zfp521 may also modulate other regulatory mechanisms that control chondrocyte proliferation and apoptosis. In the absence of Zfp521, the organization of the growth plate is disturbed and PTHrP responses are altered. Thus, Zfp521 is an important regulator of growth plate differentiation and endochondral bone development that is required for key downstream responses to PTHrP.
Experimental Procedures
Cell and Organ Cultures
ATDC5 cells were expanded and cultured in micromass as previously described (Shukunami et al., 1996). The medium was changed every other day and supplemented with PTHrP (1-34), PTHrP (7-34), forskolin, H-89 and TPA as indicated. MCT cells were cultured as previously described (Zheng et al., 2003). PTHrP (1-34) or forskolin were added to the cultures at the beginning of the hypertrophic differentiation (non-permissive temperature). Primary chondrocytes were isolated from rib cages of mice, cultured as previously described (Lefebvre et al., 1994), and treated with PTHrP (1-34) or PTHrP (7-34) for 48 hours. Metatarsals from 2 week-old mice hind limbs were cultured in α-MEM medium supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), and 1% L-glutamine with or without PTHrP (1-34) for 10 days in 37 °C/5% CO2, changing medium every other day. MC3T3-E1 cells were cultured as previously described (Wu et al., 2009). NIH3T3 cells were cultured in DMEM medium supplemented with 10% BCS, penicillin (100 U/ml), streptomycin (100 μg/ml).
Gene reporter assay
MCT cells (2 × 105 cells/well in six-well plates) were cultured at 32 °C (permissive temperature) until 70% confluent. Cells were transfected according to manufacturer's instructions using FuGene6 (Roche) in a 3:1 ratio with the 6xOSE2-Luc reporter plasmid along with combinations of Flag-Runx2 (0.5μg), HA-Zfp521 (1 μg), HDAC4 siRNA (0.5μg) and GAPDH siRNA (0.5ug) as indicated and adjusting the total amount of transfected DNA with empty vector (pcDNA 3.1). Zfp521 dose response was assayed using Zfp521:Runx2 ratios of 1:1, 3:1 and 5:1. pCMV-Renilla-luciferase (Promega) (17ng) was used for normalization. Transfected cells were cultured for 48 h at 37 °C/5% CO2 (non-permissive temperature) and luciferase activity was determined using the dual-luciferace reporter assay kit (Promega) following manufacturer's instructions. Results were calculated as fold activation over empty vector control.
Co-Immunoprecipitation assay
Co-immunoprecipitation was performed essentially as described elsewhere (Purev et al., 2009) using 2 μg mouse monoclonal anti-HDAC4 (Santa Cruz), 2 μg rabbit polyclonal anti-Zfp521 (Wu et al., 2009), or 2 μg anti-mouse IgG antibody (Promega) and Protein A/G-agarose bead slurry (Santa Cruz). Beads were collected by centrifugation and washed 3-4 times in mRIPA. The immune complexes were boiled in 2× SDS-PAGE sample buffer and subjected to immunoblot analysis.
Tet-inducible Zfp521 overexpression system
ATDC5 cells stably transfected with Tet-inducible Zfp521 were generated using the T-Rex inducible system (Invitrogen) according to manufacturer's instructions. Doxycycline (0.5 μg/ml) was added to culture medium to induce Zfp521 overexpression. ATDC5 cells stably transfected with Tet-inducible LacZ were used as control. mRNA was assessed by qPCR.
Zfp521 shRNA-expressing cells
Two ATDC5 cell lines that expressed Zfp521 shRNAs were created using MISSION® shRNA lentiviral transduction particles (Sigma-Aldrich) encoding five different shRNA sequences (Broad Institute, Cambridge, MA) (protocol available at www.sigmaaldrich.com). ATDC5 cells were transduced at 60% confluence (MOI 2 and 5) according to manufacturer's instructions and selected with puromycin (4 μg/m). Knockdown efficiency was evaluated by measuring Zfp521 expression in 12 day micromass cultures by qPCR. Cells expressing either non-target shRNA or TurboGFP control shRNAs were prepared similarly. MC3T3-E1 cells stably expressing non-target shRNA or Zfp521 shRNA (seq. #4) were established similarly.
Generation of a chondrocyte-specific Zfp521 conditional knockout mouse
The Zfp521 gene was specifically deleted from chondrocytes by crossing floxed Zfp521 (Kiviranta et al., manuscript in preparation) with mice that express the Cre recombinase under the control of the collagen type II (α1) promoter (Long et al., 2001).
Growth Plate histology and histomorphometry
Hind limbs from 2 week-old Zfp521 cKO and floxed control mice were dissected and processed for paraffin and methylmethacrylate embedding with and without decalcification, respectively, as previously described (Wu et al., 2009). Paraffin sections were stained with Safranin O. von Kossa staining of mineralized matrix was performed on methylmetacrylate sections after plastic removal and analyzed under an inverted microscope (Maeda et al., 2007). Growth plate histomorphometry was performed on Safranin O-stained sections of proximal tibia growth plates using Osteomeasure software (Osteometrics–Decatur). A representative field at 40× was used to obtain cell counts, mean polar cellular axes (cell heights) and stereological volumetric estimators obtained from measured areas (volume occupied by ECM and cells respectively in the hypertrophic zone) as described by Hunziker (Hunziker et al., 1987) with slight modifications. Briefly, cumulative hypertrophic chondrocyte surface area was measured and subtracted from the total hypertrophic zone surface area of the field obtaining the ECM area component of the zone. The area measurements served as volumetric stereological estimators of respective volumes.
Proliferation assays
Two week-old mice were injected intraperitoneally with 200 μl (1 ml/100 g of body weight) of BrdU labeling reagent (Zymed laboratories) 20 and 4 hours before sacrifice. Incorporated BrdU was localized in proximal tibiae by immunohistochemistry using the Cell Proliferation IHC kit, BrdU detection, with DAB visualization (Chemicon) according to manufacturer's instructions. Zfp521 shRNA-expressing ATDC5 cells and Tet-inducible Zfp521 ATDC5 cells were cultured on coverslips for 7 days and exposed to BrdU labeling reagent for 14 hours before fixing in 3.7% formaldehyde for 10 min and BrdU immunolocalization. BrdU incorporation was quantified using the Cell Proliferation ELISA kit, BrdU detection (Chemicon). MC3T3-E1 cells stably expressing non-target or Zfp521 shRNA and NIH3T3 cells transiently expressing empty vector (pCMV2) or Zfp521 (pCMV2-Zfp521) were cultured on coverslips for 24 hours and exposed to BrdU labeling reagent for 14 hours before BrdU immunolocalization. Percentage of BrdU-positive cells was determined.
Caspase activity and TUNEL assay
105 primary rib chondrocytes were cultured on coverslips for 5 days. 105 shRNA-expressing ATDC5 cells or 105 ATDC5 cells stably transfected with Tet-inducible Zfp521 were cultured on coverslips in differentiation medium with or without doxycycline (0.5 μg/ml), respectively, for 1 week. Caspase activity was assessed by fluorescence microscopy using the Sulforhodamine FLICA Apoptosis Detection Kit Caspase Assay (Immunochemistry-Bloomington) according to manufacturer's instructions. TUNEL labeling was done using an In Situ Death Detection Kit, Fluorescein (Roche) on paraffin-embedded proximal tibiae sections, following manufacturer's instructions. The number of apoptotic cells was quantified.
In Situ Hybridization (ISH)
35S-UTP-labelled riboprobes were synthesized from linearized plasmids using the in vitro transcription kit (Promega) and 35S-UTP (Amersham). ISH using antisense riboprobes was performed on 3.7% formaldehyde-fixed, decalcified, paraffin-embedded proximal tibias of 2 week old mice and metatarsal bones as previously described (Razzaque et al., 2005). Image J 1.42q software (Wayne Rasband, National Institutes of Health, USA) was used to calculate growth plate thicknesses and distances, measured in arbitrary units and reported as percentage changes, and the granular area fraction covered by the ISH signal of Col X, PTHR1 and Ihh as a measure of amount of signal, and reported as fold-change.
Statistical Analysis
Statistical differences were assessed with the unpaired Student's t test. Data are presented as mean ± SD, and p values smaller than 0.05 were considered statistically significant.
Additional information about antibodies, transient transfections, quantitative PCR, Western blotting, Immunohistochemistry, and Alcian Blue/Alizarin Red staining is included in the supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Dr. Brendan Lee for MCT cells, Dr. Ernestina Schipani for the PTHR1 – HKrk-H223R transgenic mice, Dr. Arthur Broadus for the PTHrP-/- mice, Dr. Andrew Lassar for HDAC4 and GAPDH siRNA plasmids, Dr. Fanxin Long for the Col II-Cre mice, and Dr. Henry Kronenberg for helpful discussion. Support was provided in part by National Institutes of Health-NIAMS grants AR048218 (R.B.) and AR050560 (B.L.). Additional support was provided by NIH training grant GM07527 (D.C.), the George Robert Pfeiffer Fellowship from the Gustavus and Louise Pfeiffer Research Foundation (D.C.), the Deutsche Forschungsgemeinschaft (E.H., HE 5208/1-1), the Gideon & Sevgi Rodan Fellowship from the International Bone and Mineral Society (E.H. and R.K.), the Academy of Finland (R.K.), and the Harvard School of Dental Medicine Dean's Scholars Award (E.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amizuka N, Davidson D, Liu H, Valverde-Franco G, Chai S, Maeda T, Ozawa H, Hammond V, Ornitz DM, Goltzman D, et al. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone. 2004;34:13–25. doi: 10.1016/j.bone.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Amizuka N, Henderson JE, Hoshi K, Warshawsky H, Ozawa H, Goltzman D, Karaplis AC. Programmed cell death of chondrocytes and aberrant chondrogenesis in mice homozygous for parathyroid hormone-related peptide gene deletion. Endocrinology. 1996;137:5055–5067. doi: 10.1210/endo.137.11.8895380. [DOI] [PubMed] [Google Scholar]
- Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126:1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Beier F, Ali Z, Mok D, Taylor AC, Leask T, Albanese C, Pestell RG, LuValle P. TGFbeta and PTHrP control chondrocyte proliferation by activating cyclin D1 expression. Mol Biol Cell. 2001;12:3852–3863. doi: 10.1091/mbc.12.12.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Leask TA, Haque S, Chow C, Taylor AC, Lee RJ, Pestell RG, Ballock RT, LuValle P. Cell cycle genes in chondrocyte proliferation and differentiation. Matrix Biol. 1999;18:109–120. doi: 10.1016/s0945-053x(99)00009-8. [DOI] [PubMed] [Google Scholar]
- Bond HM, Mesuraca M, Amodio N, Mega T, Agosti V, Fanello D, Pelaggi D, Bullinger L, Grieco M, Moore MA, et al. Early hematopoietic zinc finger protein-zinc finger protein 521: a candidate regulator of diverse immature cells. Int J Biochem Cell Biol. 2008;40:848–854. doi: 10.1016/j.biocel.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Bond HM, Mesuraca M, Carbone E, Bonelli P, Agosti V, Amodio N, De Rosa G, Di Nicola M, Gianni AM, Moore MAS, et al. Early hematopoietic zinc finger protein (EHZF), the human homolog to mouse Evi3, is highly expressed in primitive human hematopoietic cells. Blood. 2004;103:2062–2070. doi: 10.1182/blood-2003-07-2388. [DOI] [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, Yayon A, Deng CX. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, Kishimoto T, Komori T. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275:8695–8702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, Kronenberg HM. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol. 2006;292:116–128. doi: 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB, Schenk RK, Cruz-Orive LM. Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone growth. J Bone Joint Surg Am. 1987;69:162–173. [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Kitagaki J, Tamamura Y, Gentili C, Koyama E, Enomoto H, Komori T, Pacifici M, Enomoto-Iwamoto M. Runx2 expression and action in chondrocytes are regulated by retinoid signaling and parathyroid hormone-related peptide (PTHrP) Osteoarthritis Cartilage. 2003;11:6–15. doi: 10.1053/joca.2002.0860. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Morse HC, 3rd, Jenkins NA, Copeland NG. Identification of Evi-3, a novel common site of retroviral integration in mouse AKXD B-cell lymphomas. J Virol. 1994;68:1293–1300. doi: 10.1128/jvi.68.3.1293-1300.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Alliston T, Delston R, Derynck R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24:2543–2555. doi: 10.1038/sj.emboj.7600729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986. doi: 10.1242/dev.129.12.2977. [DOI] [PubMed] [Google Scholar]
- Kozhemyakina E, Cohen T, Yao TP, Lassar AB. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol. 2009;29:5751–5762. doi: 10.1128/MCB.00415-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM. PTHrP and skeletal development. Ann N Y Acad Sci. 2006;1068:1–13. doi: 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- La Rocca R, Fulciniti M, Lakshmikanth T, Mesuraca M, Ali TH, Mazzei V, Amodio N, Catalano L, Rotoli B, Ouerfelli O, et al. Early hematopoietic zinc finger protein prevents tumor cell recognition by natural killer cells. J Immunol. 2009;182:4529–4537. doi: 10.4049/jimmunol.0802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J Clin Invest. 1999;104:399–407. doi: 10.1172/JCI6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Garofalo S, Zhou G, Metsaranta M, Vuorio E, De Crombrugghe B. Characterization of primary cultures of chondrocytes from type II collagen/beta-galactosidase transgenic mice. Matrix Biol. 1994;14:329–335. doi: 10.1016/0945-053x(94)90199-6. [DOI] [PubMed] [Google Scholar]
- Li TF, Dong Y, Ionescu AM, Rosier RN, Zuscik MJ, Schwarz EM, O'Keefe RJ, Drissi H. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp Cell Res. 2004;299:128–136. doi: 10.1016/j.yexcr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229:141–162. doi: 10.1006/dbio.2000.9975. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, Mackem S, Lanske B. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104:6382–6387. doi: 10.1073/pnas.0608449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara E, Sakai I, Yamanouchi J, Fujiwara H, Yakushijin Y, Hato T, Shigemoto K, Yasukawa M. The role of zinc finger protein 521/early hematopoietic zinc finger protein in erythroid cell differentiation. J Biol Chem. 2009;284:3480–3487. doi: 10.1074/jbc.M805874200. [DOI] [PubMed] [Google Scholar]
- Nutt RF, Caulfield MP, Levy JJ, Gibbons SW, Rosenblatt M, McKee RL. Removal of partial agonism from parathyroid hormone (PTH)-related protein-(7-34)NH2 by substitution of PTH amino acids at positions 10 and 11. Endocrinology. 1990;127:491–493. doi: 10.1210/endo-127-1-491. [DOI] [PubMed] [Google Scholar]
- Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Purev E, Neff L, Horne WC, Baron R. c-Cbl and Cbl-b Act redundantly to protect osteoclasts from apoptosis and to displace HDAC6 from {beta}-tubulin, stabilizing microtubules and podosomes. Mol Biol Cell. 2009;20:4021–4030. doi: 10.1091/mbc.E09-03-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS, Soegiarto DW, Chang D, Long F, Lanske B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J Pathol. 2005;207:453–461. doi: 10.1002/path.1870. [DOI] [PubMed] [Google Scholar]
- Schipani E, Jensen GS, Pincus J, Nissenson RA, Gardella TJ, Juppner H. Constitutive activation of the cyclic adenosine 3′,5′-monophosphate signaling pathway by parathyroid hormone (PTH)/PTH-related peptide receptors mutated at the two loci for Jansen's metaphyseal chondrodysplasia. Mol Endocrinol. 1997;11:851–858. doi: 10.1210/mend.11.7.9934. [DOI] [PubMed] [Google Scholar]
- Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with Runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004;279:41998–42007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- Shukunami C, Shigeno C, Atsumi T, Ishizeki K, Suzuki F, Hiraki Y. Chondrogenic differentiation of clonal mouse embryonic cell line ATDC5 in vitro: differentiation-dependent gene expression of parathyroid hormone (PTH)/PTH-related peptide receptor. J Cell Biol. 1996;133:457–468. doi: 10.1083/jcb.133.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto-Iwamoto M, Ohmori T, Enomoto H, Nakata K, Takada K, et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. 2001;153:87–100. doi: 10.1083/jcb.153.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Warming S, Liu P, Suzuki T, Akagi K, Lindtner S, Pavlakis GN, Jenkins NA, Copeland NG. Evi3, a common retroviral integration site in murine B-cell lymphoma, encodes an EBFAZ-related Kruppel-like zinc finger protein. Blood. 2003;101:1934–1940. doi: 10.1182/blood-2002-08-2652. [DOI] [PubMed] [Google Scholar]
- Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf JJ, Zaidi SK, Cascino JE, Kahler R, van Wijnen AJ, Lian JB, Yoshida M, Stein GS, Li X. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol Cell Biol. 2002;22:7982–7992. doi: 10.1128/MCB.22.22.7982-7992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hesse E, Morvan F, Zhang JP, Correa D, Rowe GC, Kiviranta R, Neff L, Philbrick WM, Horne WC, et al. Zfp521 antagonizes Runx2, delays osteoblast differentiation in vitro, and promotes bone formation in vivo. Bone. 2009;44:528–536. doi: 10.1016/j.bone.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka Y, Tanaka H, Koike M, Nishimura R, Seino Y. PTHrP rescues ATDC5 cells from apoptosis induced by FGF receptor 3 mutation. J Bone Miner Res. 2003;18:1395–1403. doi: 10.1359/jbmr.2003.18.8.1395. [DOI] [PubMed] [Google Scholar]
- Yamasaki N, Miyazaki K, Nagamachi A, Koller R, Oda H, Miyazaki M, Sasaki T, Honda ZI, Wolff L, Inaba T, et al. Identification of Zfp521/ZNF521 as a cooperative gene for E2A-HLF to develop acute B-lineage leukemia. Oncogene. 2010;29:1963–1975. doi: 10.1038/onc.2009.475. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.