

Abstract
Functional selectivity is the term that describes drugs that cause markedly different signaling through a single receptor (e.g., full agonist at one pathway and antagonist at a second). It has been widely recognized recently that this phenomenon impacts the understanding of mechanism of action of some drugs, and has relevance to drug discovery. One of the clinical areas where this mechanism has particular importance is in the treatment of schizophrenia. Antipsychotic drugs have been grouped according to both pattern of clinical action and mechanism of action. The original antipsychotic drugs such as chlorpromazine and haloperidol have been called typical or first generation. They cause both antipsychotic actions and many side effects (extrapyramidal and endocrine) that are ascribed to their high affinity dopamine D2 receptor antagonism. Drugs such as clozapine, olanzapine, risperidone and others were then developed that avoided the neurological side effects (atypical or second generation antipsychotics). These compounds are divided mechanistically into those that are high affinity D2 and 5-HT2A antagonists, and those that also bind with modest affinity to D2, 5-HT2A, and many other neuroreceptors. There is one approved third generation drug, aripiprazole, whose actions have been ascribed alternately to either D2 partial agonism or D2 functional selectivity. Although partial agonism has been the more widely accepted mechanism, the available data are inconsistent with this mechanism. Conversely, the D2 functional selectivity hypothesis can accommodate all current data for aripiprazole, and also impacts on discovery compounds that are not pure D2 antagonists.
Keywords: functional selectivity, antipsychotic drugs, schizophrenia, receptor mechanisms, G protein-coupled receptors, dopamine receptors, serotonin receptors, drug action
A. The first two generations of antipsychotic drugs
The serendipitous observations about antipsychotic actions of chlorpromazine [1] ultimately led to the finding that these antipsychotic effects were due to antidopaminergic actions [2]. Numerous antipsychotics were subsequently developed that, like chlorpromazine, work primarily as dopamine D2 receptor antagonists. Drugs of this type (first called typical and now named first generation antipsychotics) include a variety of different chemical classes. These typical drugs can be classified as high, intermediate, or low potency based on their average daily dose range, with their clinical potency often highly correlated with their affinity for the dopamine D2 receptor [3]. Thus, actions at dopamine systems have been a critical mechanism in all currently approved antipsychotic drugs and many of the compounds in the discovery and development pipeline.
1. Dopamine systems and their receptors
There are three major brain dopamine pathways: the nigrostriatal (from cells in the A9 region); the mesolimbic and mesocortical systems (from cells in the A10 or ventral tegmentum); and the tuberoinfundibular (hypothalamic) system [4]. The first evidence for a biochemical mechanism of dopamine neurotransmission was the observation that dopamine could dosedependently stimulate the synthesis of the second messenger cAMP [5] in a fashion that was antagonized by typical antipsychotic drugs [6]. Both phenothiazine and thioxanthene antipsychotics competitively inhibited the dopamine-stimulated activity of adenylate cyclase in proportion to their clinical potency, leading to the first hypothesis that this was the major functional mechanism of dopamine in the CNS [6, 7]. Studies with antipsychotics in other structural families, however, showed that some behaviorally potent antipsychotics had little potency in inhibiting dopamine-stimulated adenylate cyclase [8]. This discrepancy led to the hypothesis that there were multiple dopamine receptors, one of which stimulated adenylate cyclase [5], whereas the other bound antipsychotics with affinity proportional to clinical antipsychotic potency, but whose functional effect was then unknown [3]. This bifurcation [9, 10] has been codified in the use of the terms D1-like and D2-like receptors, respectively.
These two pharmacological families are now known to be the products of five genes, and their biology has been the subject of many chapters and books [11-14]. Both of the “D1-like” receptor genes are intron-less, and include the D1 (also called the D1A in murine species) and the D5 (also termed D1B) [15-18]. The “D2-like” receptors include two major splice variants of the D2 gene, D2L (long), and D2S (short), that together are the most highly expressed of the D2-like receptors [19-22]. The other D2-like receptors are D3 and D4 receptors, and include D3 receptor splice variants that are expressed in mice but not humans [20, 23]. The D4 receptor has variable 16-nucleotide repeats in the region coding for the third intracellular loop that can vary between individuals in periodicity from 4 to 16 repeats [24, 25]. It has been suggested that these D4 alleles have major effects on behavioral phenotype [26-28], although this idea is controversial [29-31].
In addition to schizophrenia, there has been a great interest of dopamine neurotransmission in the treatment and or etiology of many central disorders such as Parkinson's disease, ADHD, etc. For this reason, there have been major efforts to discover and develop drugs that target dopamine receptors. This has resulted in a rich availability of clinical and research ligands that has facilitated basic research, and has led to many interesting clinical hypotheses.
2. Actions of first generation antipsychotics
After the accidental discovery of chlorpromazine, many other phenothiazine analogs (e.g., fluphenazine, thioridazine, etc., Figure 1) followed it to market. In addition, other first generation drugs were derived from distinct chemical backbones, such as the butyrophenone haloperidol and the thioxanthene flupenthixol (Figure 1).
Figure 1.
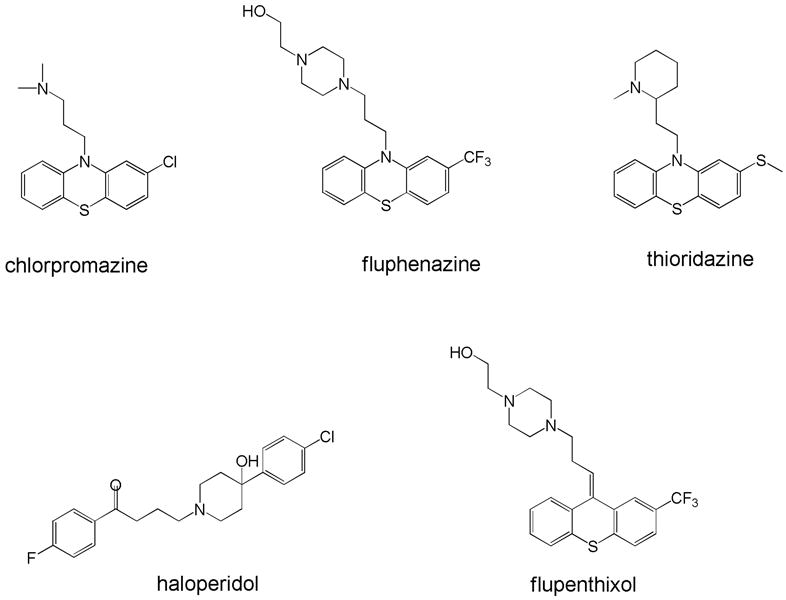
Structures of representative first generation (typical) drugs. The top row is representative of the many diverse phenothiazines that have been marketed, whereas the bottom row shows the widely used butyrophenone haloperidol and a representative thioxanthene, flupenthixol.
The widely used first generation antipsychotics like haloperidol are effective in many patients in attenuating the positive symptoms of schizophrenia (e.g., hallucinations and delusions) associated with acute episodes, as well as preventing psychotic relapse [32]. These agents have, however, important limitations. As many as 25-60% of patients are either largely resistant, or only partially responsive, to treatment with conventional antipsychotics. Moreover, these drugs at best have only a modest benefit on negative symptoms (e.g., blunted affect, poverty of thought, emotional and social withdrawal, and lack of drive). The first generation antipsychotics also induce a variety of adverse effects both acutely and with long term exposure. Such side effects reduce compliance and represent major limitations of the drugs for adequate treatment of schizophrenia. Some of the side effects (sedation, autonomic and cardiovascular effects, weight gain) are largely due to off-site actions at other receptors.
These drugs also induced a variety of neurological adverse effects seen both acutely [extrapyramidal side effects (EPS) such as parkinsonism, dystonia, akathisia, as well as neuroendocrine effects] and with long term exposure [e.g., tardive dyskinesia (TD)] [33]. These latter effects seem mediated by the same receptors (dopamine D2-like) that appear to cause therapeutic effects. Indeed, a majority of patients given recommended therapeutic dosages of typical antipsychotics develop acute EPS [34]. The more worrisome neurological side effect, tardive dyskinesia (TD), can be irreversible, and incidence rates were estimated at about 5% per year [35]. Anticholinergic drugs are effective in attenuating EPS, but have their own unpleasant side effects (e.g. dry mouth, constipation, delirium and memory deficits), further contributing to treatment non-compliance.
Worse, negative symptoms, mood symptoms, and cognitive deficits are only minimally responsive to conventional antipsychotics. The presence of negative symptoms and cognitive impairment often leads to poor social and vocational function [36, 37]. Moreover, approximately 30% of patients with acutely exacerbated psychotic symptoms have minimal response to standard antipsychotics [35, 38]. These factors spurred research for new drugs that would have equal or better efficacy, and markedly decreased side effects. For more than a decade, the side effects that were considered most critical were the neurological (EPS and TD) and neuroendocrine. The first drugs devoid of neurological side effects were originally termed “atypical” antipsychotics, but are now often called second generation.
3. Second generation antipsychotics (the “atypicals”)
For many years there was the common view that effective antipsychotic agents would necessarily produce EPS because these actions were mediated by blockade of the same receptor (i.e., D2). A great deal of research was devoted to the discovery of drugs that were “atypical” -- although there was no convention about the meaning of the term “atypical.” In its broadest sense, it was used to refer to drugs that had at least equal antipsychotic efficacy to the “typical” drugs, without producing EPS or sustained prolactin elevation [39]. With time and after the development of drugs that could be called “atypical,” the definition was often expanded to include compounds that might have superior antipsychotic efficacy (e.g., in treatment resistant patients) or have beneficial effects against negative symptoms and/or cognitive deficits.
The first drug that was sometimes called atypical was thioridazine (including its metabolite mesoridazine that was also marketed separately). Although once considered the prototypical atypical antipsychotic [40], thioridazine appears to be a moderate affinity antidopaminergic that was different largely because of its intrinsic antimuscarinic affinity [41-43]. There were no major advantages over the concomitant use of a titered dose of an anticholinergic in combination with other first generation drugs. It was the two other types of drugs that were developed that formed the backbone of what became the true atypical antipsychotics, and now make up what are termed second generation drugs. The most remarkable of these was clozapine.
a. “Rich” pharmacology: clozapine and related drugs
Clozapine underwent extensive clinical testing in the 1970's, but its development was halted in the United States and limited in other countries because of the incidence of the potentially fatal side effect, agranulocytosis. Yet clozapine was reborn because it was recognized that it was superior to traditional antipsychotics such as haloperidol and chlorpromazine in treatment of refractory schizophrenia [44]. Clozapine has little liability to induce EPS or TD, and causes far less of an increase in prolactin concentration relative to typical antipsychotics [45]. It has also been suggested that it may ameliorate (or at least not worsen) some of the negative symptoms and cognitive deficits in schizophrenia [46]. Since its re-introduction, the unparalleled clinical efficacy of clozapine has been widely recognized such that it is used despite the need for slow titering and for monitoring for blood dyscrasias. Indeed, this led to broad efforts to both understand the mechanisms that make clozapine unique, and to develop drugs that retain its remarkable efficacy but have fewer side effects.
Clozapine possesses many characteristics that distinguish it from the typical antipsychotics [37, 46]. The drug has modest affinity for some dopamine receptors (DARs) (D4, as well as D2 and D1), but also has affinity for serotonin (5-HT) 5-HT2A, H1 histaminergic, muscarinic, and α-adrenergic receptors [47]. Olanzapine has both structural and pharmacological similarity to clozapine, and has affinity for 5-HT2A, D2, as well as other dopamine, serotonin, adrenergic, and histamine receptors. Quetiapine and ziprasidone differ somewhat from clozapine in relative affinities for dopamine and a few of these “off-site” targets [48]. The clinical significance of such differences sometimes appears clear (e.g., the 5-HT1A partial agonist properties of ziprasidone may contribute to low EPS). Yet with such a diverse pharmacological profile, it is not surprising there has been controversy about which pharmacological actions of clozapine are responsible for its clinical efficacy. Because of the needed dose titering, significant occurrence of agranulocytosis and seizures, as well as sedation, hypotension, hypersalivation, and weight gain, there was a broad search for improved drugs that resulted in the marketing of olanzapine, quetiapine, and ziprasidone. There have been numerous reviews [36, 37, 49] of the pharmacology of these agents, all of which share the property of having affinity for numerous receptors that might be related to antipsychotic action and/or side effects. Elucidation of a verifiable single mechanism has escaped the field, and indeed led to the suggestion that the unique actions of clozapine were a result of multi-receptor targeting. Because the exact nature of the multiple receptor actions is critical but not well understood, it has been suggested that the term “dirty” drug should be supplanted by the euphemism “rich” pharmacology [49]. Other drugs with “rich” pharmacology like olanzapine, quetiapine, and ziprasidone are clearly effective and do not have the hematologic side effects of clozapine, but none quite matches the efficacy of clozapine [50]. Thus, the question of mechanism (is it a mixture of receptor actions or a unique effect at one of the target receptors?) is more than of heuristic interest.
b. Dopamine-serotonin antagonists
The second generation drugs include compounds that also work somewhat differently than clozapine. Unlike the relatively low D2 and 5-HT2A affinity of clozapine, ziprasidone, and quetiapine, this mechanism involves high affinity blockade of both serotonin 5-HT2A and dopamine D2 receptors [51, 52]. Antipsychotics with markedly lower propensity to cause EPS and TD are thought to work by “serotonin-dopamine antagonism”. The prototype of such drugs is risperidone (see Figure 2), although there are several structurally and mechanistically-related compounds approved or in the pipeline. Besides being a high affinity 5-HT2A and D2 antagonist, risperidone is relatively devoid of anticholinergic activity and has lower affinity for α-adrenergic and histamine H1 receptors [53]. At high doses, risperidone causes some dose-related EPS (> 6 mg/day), but the risk of neurological side effects is modest at lower doses [54]. Risperidone causes hyperprolactinemia by virtue of its blockade of D2 receptors, and is associated with modest weight gain that is a side effect of many first and second generation drugs.
Figure 2.
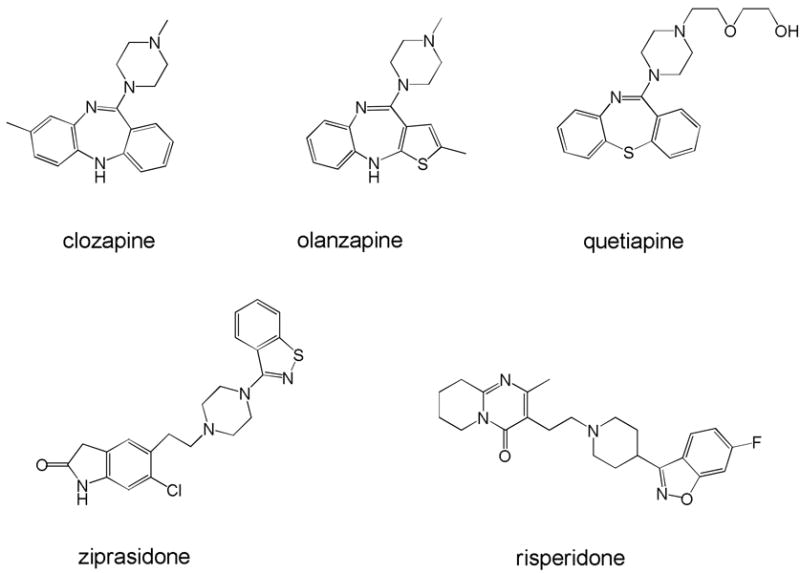
Structures of representative second generation drugs. The top row shows clozapine and two compounds in which the structural similarity can be easily seen. The bottom row shows the structure of ziprasidone, a fourth compound with “rich” pharmacology, as well as risperidone, the prototype of the “DA-5-HT potent dual antagonist.”
B. Third generation antipsychotics: partial agonists or something more?
1. Auto- and presynaptic dopamine receptors as antipsychotic candidates
One of the major driving forces for antipsychotic drug discovery focusing on the D2 receptor was the “dopamine hypothesis of schizophrenia” which, in its 1980's rendition, posited that excess dopamine release and/or excess sensitivity of dopaminoreceptive neurons, was the cause of what are now called positive symptoms. Although antagonism of D2 receptors has been accepted as a way of controlling positive symptoms of schizophrenia [3], a novel hypothesis was developed in the late 1970's. Dopamine autoreceptors (now known to be composed of high densities of D2 and low densities of D3 receptors) [11]. When activated, they cause a decrease in both synthesis and release of dopamine, and a decrease in the firing of dopamine neurons [11]. Interestingly, both dopamine and D2 agonists tend to have higher potency at these autoreceptors versus what are regarded as postsynaptic D2 functions [55]. This would then explain why the behavioral response to a full D2 agonist is typically biphasic with respect to dose, with inhibition seen at in low doses (the result of autoreceptor stimulation) and stimulation at higher doses (direct postsynaptic activation). One of the major mechanisms explaining this biphasic effect is greater presynaptic D2-like receptor reserve [56].
These observations led to the apparently contradictory idea of also using D2-like agonists to cause antipsychotic effects via one of two alternative mechanisms. One was to use a low dose of a full agonist to achieve selective presynaptic activation, the other to use a partial agonist. According to pharmacological theory, a partial agonist should be far more efficacious at activating the presynaptic receptors where there is high receptor reserve [57]. Indeed, a plethora of in vitro and animal studies suggested that the partial agonist, (-)3-PPP (preclamol), might be an excellent candidate. Although both mechanisms have a sound theoretical basis, the early clinical data were disappointing [58-63]. In retrospect, this may have reflected issues such as getting the “right” presynaptic relative receptor occupancy without temporal fluctuations, or finding a partial agonist with just the “right” intrinsic activity. On the other hand, there is a widely-held belief that a partial agonist with the “just right” degree of partial agonism has now been found, aripiprazole.
2. Aripiprazole as the third generation antipsychotic prototype
Aripiprazole is a relatively new approved antipsychotic drug proffered by its developers as a high affinity, low intrinsic activity partial D2 agonist. Although the compound has effects on several other receptors, many of the leading figures in schizophrenia biology have labeled aripiprazole as the first “dopamine stabilizer” based on these purported D2 partial agonist properties [64-66]. According to this view, in situations of high extracellular dopamine concentrations (e.g., in mesolimbic areas involved in positive symptoms), the partial agonist properties of aripiprazole compete with dopamine and cause partial antagonism offering clinical benefit. Conversely, in situations where extracellular dopamine concentrations are low (for example in dopamine circuits involved in working memory), the drug can occupy additional receptors and cause partial activation. A cartoon indicating how this is proposed to occur is shown in Figure 3. On its face, this seems to provide a logical and cogent mechanism that combines classic pharmacological logic about mechanisms of partial agonism with recent information about the biology of schizophrenia.
Figure 3.

Cartoon illustrating how the proposed D2 dopamine partial agonist mechanism works in third generation antipsychotics. Left column: mesolimbic dopaminergic transmission. Right column: Prefrontal cortical dopaminergic transmission. Broad dotted line: “normal” dopamine activity; solid dark box: Abnormal transmission in schizophrenia; Solid sigmoidal line: actions of partial agonist alone. Dotted sigmoidal line: actions of partial agonist in the presence of endogenous concentrations of dopamine.
In some functional assay systems, aripiprazole is indeed a low-to-moderate intrinsic activity partial agonist [67-69] as required by this prevalent hypothesis. On the other hand, much of the available data with aripiprazole are problematic as it relates to this partial agonist hypothesis. The intrinsic activity and potency of aripiprazole for the D2-mediated inhibition of cAMP accumulation is cell line-dependent. Thus, the drug has weak partial agonist activity in the CHO-D2L cell line, but strong partial agonist activity in HEK-D2L cells [67-69]. In addition, aripiprazole has markedly different potencies at two D2L-mediated functions within the same cell line [70], or even at the same function in two different cell lines [69]. Moreover, aripiprazole is a pure antagonist at both D2 agonist-mediated GTPγS binding and GIRK channel activity [69], whereas it is a full agonist in situ for D2-mediated inhibition of tyrosine hydroxylase [71]. Thus aripiprazole appears to elicit D2-mediated functional effects that encompass the whole range of classic pharmacological intrinsic activity. This degree of functional discrimination is not seen with other “partial agonists.” These large variations in both intrinsic activity and potency, not explicable by other mechanisms, that have led to the hypothesis that aripiprazole is “functionally selective” [67, 69, 72] at D2 receptors, not just a simple partial agonist. It is now clear that “functional selectivity” is not a mechanism unique for aripiprazole.
3. Functional selectivity and the notion of intrinsic efficacy
For the last half century, pharmacological theory has posited that ligands could be characterized by the nature of the functional effects elicited by their interaction with their target receptors [73]. These effects are governed by two important properties: affinity, the property of attraction between a ligand and its receptor, and efficacy, the property that allows ligands, once bound, to produce a response [74]. This concept has led to the classification of a drug as a full agonist, a partial agonist, a neutral antagonist, or an inverse agonist. The notion was that each drug had innate “intrinsic efficacy,” a measure of the stimulus per receptor molecule produced by a ligand at its target receptor [75]. A corollary of this idea was that the ability of a ligand to impart (or reduce) stimulus once that ligand is bound to the receptor is an inherent property of the ligand-receptor complex. Intrinsic efficacy is thus differentiated from the more operational term “intrinsic activity” (Ariens, 1954) that refers to the maximal effect (Emax) of a ligand relative to a reference agonist in any single given experimental system. Historically, pharmacologists have viewed intrinsic efficacy as a system-independent parameter that is constant for each ligand at a given receptor, irrespective of where the target receptor is expressed. Differences in the degree of agonism, for example, in different tissue types were assumed to reflect differences in receptor density and/or the strength of stimulus-response coupling. This classification of drug action based on intrinsic efficacy allowed for variations in the quantity of the stimulus that was imparted to the cell, but not the quality. Thus, a full agonist would be expected to activate all of the signaling pathways linked to a receptor to the same degree as the endogenous ligand for that receptor. During the past 15 years, however, a huge body of data has emerged suggesting that the premise of “intrinsic efficacy” may be invalid.
4. Early evidence for functional selectivity of dopaminergic compounds
To our knowledge, the first clear example of dopamine receptor functional selectivity resulted from serendipitous findings with dihydrexidine, a compound originally designed to be a selective D1 agonist [76]. After dihydrexidine was found to have D2 affinity [77, 78], both it and a more D2 selective analog N-n-propyldihydrexidine (propylDHX) were characterized functionally. From both binding and canonical functional assays, both compounds appeared to be full D2 agonists. They competed for D2 receptors in heterologous systems or in brain tissue with shallow, guanine-nucleotide-sensitive curves similar to typical agonists [78-80], and were full agonists at D2 receptor mediated inhibition of adenylate cyclase [79, 80]. Consistent with this, they also inhibited prolactin secretion in vivo.
Unexpectedly however, dihydrexidine and propylDHX bound to D2–like presynaptic/autoreceptors that mediate inhibition of dopamine neuron firing, dopamine release, or dopamine synthesis [79, 80], but had only antagonist properties. Off-site actions were excluded by both receptor screening and competitive pharmacological studies [78]. These agonist-antagonist actions of a single drug at a single receptor could be replicated in heterologous or single cell systems. Thus, in pituitary lactotrophs, dihydrexidine was a full agonist at D2-mediated inhibition adenylate cyclase, yet had little intrinsic activity at D2 receptors coupled to G protein-coupled inwardly-rectifying potassium channels (GIRK), and was an effective antagonist of the functional actions of dopamine [79]. In mesencephalic-derived MN9D cells transfected with the D2L receptor, both compounds were full agonists at inhibition of adenylate cyclase activity, yet were antagonists at D2L mediated inhibition of depolarization-induced release of dopamine [80]. Neither dihydrexidine nor propylDHX had effects in untransfected MN9D cells not expressing the D2L receptor.
In toto, these studies led to the formal idea, to wit, that “functional selectivity” [is] the concept that [some] drugs can cause functional multiplicity even when interacting with a single receptor isoform. …. One mechanism for functional selectivity may be atypical conformation changes [when compared to the endogenous ligand] induced when such drugs bind to the receptor-G protein complex. The particular type(s) of G-protein are dependent on both the type of cell, and the location in the cell, where the receptor of interest is located. Such functional targeting allows drug effects to be refined to a degree not possible just by targeting specific receptor isoforms. This could yield important therapeutic advances, although it introduces a new level of complexity that will require significantly greater understanding of receptor dynamics and the interaction with transduction mechanisms” [81]. This may have been the earliest conceptualization linking a speculative hypothesis [82] to actual data [81], and was soon followed by other reports of similar functional “mismatches” [see 83 for another early example].
5. Is functional selectivity an artifact or a relevant pharmacological mechanism?
For some time, the overall implications of functional selectivity to drug action in vivo was not widely accepted despite some of the available data. For example, classic pharmacology would have predicted that a compound with full intrinsic activity at both D1 and D2 receptors (i.e., dihydrexidine at both D1 and D2 receptors modulating adenylate cyclase) would have behavioral effects similar to apomorphine or amphetamine, direct or indirect activators of both families of receptor. Clearly, however, dihydrexidine did not have the expected behavioral actions of a mixed D1:D2 agonist [84]. This was consistent with the hypothesis that the atypical D2 signaling properties of the this compound markedly affected its pharmacological actions in vivo.
Equally compelling were studies done with the D2-selective analog of dihydrexidine, propyldihydrexidine (propylDHX). D2-like agonists are known to have biphasic behavioral effects, causing locomotor inhibition at low doses and locomotor stimulation and stereotypies at higher doses [85]. PropylDHX, however, only caused modest locomotor inhibition across a wide range of doses, with no competing behaviors that might have interfered with locomotion [86]. This provided additional evidence that functionally selective compounds may well have novel pharmacological actions. As we have summarized recently [72], there is a growing body of data demonstrating functional selectivity of ligands for dozens of receptors. The pharmacological impact of such differential signaling is obviously an important area of future investigation.
6. Does functional selectivity explain more of the actions of aripiprazole?
As discussed earlier, there are clear data demonstrating functionally selective properties of aripiprazole that are inconsistent with simple partial agonism [67, 69, 70]. It may be argued that the clinical effect of aripiprazole can be explained by either mechanism, leaving a clear answer currently in abeyance. Conversely, there are behavioral data that also are inconsistent with the partial agonist mechanism. Aripiprazole has been studied in the unilaterally lesioned 6-hydroxydopamine (6-OHDA) treated rat [87]. This is an extremely useful pharmacodynamic model (sometimes proposed to be a rat model of Parkinson's disease) in which both full and partial dopamine agonists cause the test animal to turn contralaterally in a tight circle towards the side away from the 6-OHDA lesion (i.e., leftward turning if the lesion was to the right-side nigrostriatal pathway). This robust rotation is a result of relative receptor/cellular hypersensitivity of the target receptors on neurons in the lesioned, dopamine-depleted, striatum. If aripiprazole was a partial agonist, it should also cause such contralateral rotation, but it does not [71]. Moreover, not only does it fail to cause rotation on its own, but it is a pure antagonist of the turning caused by known dopamine D2 agonists [71]. Here then is a situation in which aripiprazole is a pure antagonist in a system with extremely low dopamine tone. These findings are directly contradictory to what is predicted by the partial agonist hypothesis, but consistent with a functional selectivity mechanism.
Interestingly, there is some clinical lore that also may be relevant. Many Parkinson's disease (PD) patients develop psychotic side effects as a result of their use of levodopa and/or dopamine agonists (the latter largely working via D2 receptors). Clozapine and quetiapine are particularly effective in treating these side effects without worsening Parkinson's symptoms. By similar reasoning as espoused for schizophrenia, aripiprazole (as a partial agonist) also should be very useful in treating these psychotic symptoms when added to the dopaminergic regimen of the PD patient. In fact, aripiprazole not only lacks effectiveness in treating these psychoses, it tends to worsen motor function [88]. These data also are inconsistent with the partial agonist hypothesis.
Thus, the available data better support the hypothesis that aripiprazole works as a functionally selective D2 ligand where its intrinsic activity varies markedly depending on the signaling environment of the D2 receptor [67, 69, 72]. It is fascinating to hypothesize how this specifically translates into functional changes in the intact organism. It may be, for example, that the cellular changes that cause post-denervation supersensitivity also change the D2 signaling environment such that aripiprazole loses intrinsic activity. This would explain the results in the unilateral 6-OHDA rats or in PD patients. Conversely, it might be supposed that, at the critical D2 receptors in the dopamine mesolimbic terminal fields, aripiprazole is a low intrinsic activity ligand. This may explain the common observation that while well-tolerated for this class of medication, the drug may be slightly less effective than compounds with pure D2 antagonist effects.
C. Impact of functional selectivity and research issues for next generation drugs
1. Complexity of signaling through D2 receptors
It is now clear that signaling through modulation of adenylate cyclases is only a fraction of how dopamine receptors modulate cellular function [89, 90]. The first signaling pathway identified for D2-like receptors was inhibition of cAMP accumulation [91, 92], and D2 receptors inhibit adenylate cyclase in most clonal and in situ cells [93]. In situ, D2-like inhibition of adenylate cyclase can lead to presynaptic/autoreceptor decreases in tyrosine hydroxylase activity and in firing of nigrostriatal dopamine neurons. Like other Gαi/o-coupled receptors, D2-like receptors modulate many signaling pathways including phospholipases, ion channels, MAP kinases, and the Na+/H+ exchanger, as well as adenylate cyclases [94]. D2 activation has major effects on K+ currents via dissociation of Gβγ subunits. D2-like receptors activate a G protein-regulated inwardly rectifying potassium channel (GIRK or Kir3), modulating several potassium currents in various tissues [95-98]. Dopamine release-regulating autoreceptors are coupled to potassium channels [99] rather than to inhibition of adenylate cyclase [100], and there is robust regulation of GIRK currents by D2 receptors in substantia nigra dopamine neurons [101].
The D2-like receptors also affect calcium [102-106] and sodium channels [107, 108]. MAP kinases are another set of pathways that can be modulated by D2 activation [109-116] and stress-activated protein kinase/Jun amino-terminal kinase (SAPK/JNK) [111]. The D2 receptor can potentiate arachidonic acid release induced by calcium-mobilizing receptors, a response mediated by cytosolic phospholipase A2 [117-120], and also directly. The D4, but not D3, receptor also activates this pathway [120, 121]. Arachidonic acid and its lipoxygenase and cyclooxygenase metabolites have numerous effects on cellular function, including feedback regulation of D2-like signaling and dopamine transport [122-125]. Finally, the D2 receptor can also stimulate phospholipase D, catalyzing the hydrolysis of phosphatidylcholine to form choline and phosphatidic acid [126, 127].
A major mediator of D2-like signaling is the Gαi/o class of G proteins that are inactivated by pertussis toxin-catalyzed ADP-ribosylation [128, 129], but as noted earlier, non-G protein signaling has become topically important. Most evidence relates to β-arrestin2, a key member of the GPCR desensitization machinery, to mediate downstream signaling through PKB(AKT) and GSK3 [130, 131]. The evidence is based on studies using a variety of genetically altered mice that were deficient in major constituents of GPCR signaling, including GPCR kinases (GRKs), β-arrestins, the dopamine transporter (DAT), and PKB. The data showed simultaneous recruitment of both protein phosphatase 2A (PP2A) and PKB by β-arrestin2 resulting in the dephosphorylation of PKB, and the subsequent loss of the inhibitory effect of PKB on the GSK3 signaling pathway, something indispensable for neuronal survival and differentiation [130-132]. Interestingly, the time course of β-arrestin/PKB signaling at the D2 receptor takes much longer to occur (hours versus minutes) compared to the G protein-mediated cAMP/PKA response [130, 133, 134]. The slow response of the β-arrestin/PKB pathway can also be indicative of its downstream effects on gene transcription culminating in target protein transcriptional profile changes or even chromatin remodeling [135-138].
In translating findings from in vitro systems, receptor selectivity is a major confound. One complication comes from the fact that there are few, if any, drugs that are highly selective for only one of the D2-like isoforms [89, 90, 139]. Moreover, there are interesting allelic variants of the D4 receptor, and two alternative splicing-derived isoforms of the D2 receptor with different third cytoplasmic loops [i.e., D2Long (D2L) and D2Short (D2S)]. Coupled with the “off-site” actions that may very well contribute to both useful and side effects, it is not surprising that establishing an unequivocal mechanism(s) of action has been difficult.
2. Consequences of differential effects on signaling pathways mediated by a single receptor
The previous section provides an overview of how some ligands may cause markedly different patterns of signaling via occupation of D2 receptors. It is this complex matrix of signaling that we have hypothesized is a major mechanism in the unexpected behavioral effects of both experimental drugs and aripiprazole [72, 90, 140]. These manifold, often inter-related signaling patterns underscore the complexity that offers both research challenges and drug discovery opportunities. In terms of the latter, one might consider the D2 receptor signaling through canonical G protein mechanisms and for non-canonical mechanisms. The potential impact of a functionally selective ligand is illustrated in Figure 4.
Figure 4.

Cartoon illustrating how functional selectivity at a single receptor might lead to an altered balance of therapeutic and side effects [Adapted from 140]. In this example, the “typical” ligand [Left panel] is an agonist that activates two signaling pathways via single receptor. One of these pathways is specifically linked to a therapeutic effect, the other to a side effect. The “functionally selective” ligand [right panel] fully activates only the pathway linked to the therapeutic effect, thereby decreasing side effects mediated by this single receptor.
In theory, it should be possible for a ligand to discriminate among canonical functions mediated by different G protein heterotrimers, but also to cause differential longer-term responses. One of the critical challenges is how to develop high-throughput, physiologically-meaningful screening systems that can distinguish novel drug candidates. The issues inherent in such approaches are not only the assays themselves, but also the cellular background that one chooses to use. It is fair to say that functional selectivity has invigorated the interest in these issues.
3. Dopaminergic mechanisms of candidate compounds
Resolving the alternate hypotheses noted above, as well as determining the true clinical utility of functionally selective drugs, will require other compounds with functionally selective (partial agonist?) properties in vitro that are tested widely in humans. As noted above, our view is that the available data support the hypothesis that functionally selective D2 properties of aripiprazole (not D2 partial agonist effects) underlie its unique clinical profile. It is useful to briefly review some of the other compounds whose data may now, or in the future, bear on this question. The following are a few of the purported D2 partial agonists or otherwise novel D2 like ligands that may play a role in understanding these issues better (see Figure 5 for key structures).
Figure 5.

Structures of some of the mechanistically interesting compounds.
a. Compounds of specific historical or clinical importance
Preclamol [(-)-3PPP] (Figure 5) is the compound that may be considered the prototype of dopamine agonists with novel actions. The two enantiomers of 3PPP show different, sometimes opposite, effects at dopamine synaptic functions (Hjorth et al 1983). Both isomers are partial D2 agonists, with the (+) isomer having somewhat lower affinity and lower intrinsic activity partial agonist. Preclamol [(-)-3PPP] preferentially stimulates the dopamine autoreceptor, and has some limbic-selectivity as a partial D2 agonist. At higher doses it has antagonist effects at the normosensitive postsynaptic D2 site [141]. Based on these data and the view of schizophrenia illustrated in Figure 3, preclamol was tested for antipsychotic potential, but any immediate effects were not sustained with repeated dosing as occurs with accepted antipsychotic drugs [63]. Although these results did not directly support the partial agonist hypothesis, the short term benefit was taken by many as a reason to pursue this direction. Contemporaneously, Otsuka developed OPC-4392 (Figure 5), a quinolinone derivative that is an aripiprazole predecessor. It was reported to act as an agonist at presynaptic dopamine autoreceptors decreasing synthesis and release of dopamine, while acting as an antagonist at postsynaptic D2 receptors by inhibiting stereotyped and climbing behavior induced by apomorphine in mice [142-144]. Although OPC-4392 was withdrawn after early clinical trials because it worsened psychoses and increased suicidal ideation, it ultimately led to the discovery of aripiprazole. If the differences in functional selectivity between OPC-4392 and aripiprazole can be elucidated, it very well may offer clues as to the discovery of even better agents.
Substituted benzamides
The benzamides form an interesting family of dopamine receptor antagonists and potential antipsychotics whose pharmacology may well impinge on mechanisms of functional selectivity. In general, the benzamides have preferential affinity for dopamine D2-like receptors, and many of them have atypical pharmacology in brain tissue that is not completely understood. Some of the early benzamides that have been tested clinically include nemonapride (YM-09151-2) and remoxipride. Radiolabeled nemonapride is a commonly used research tool, and raclopride (FLA 870; A40664) and eticlopride (A38503; FLB131) are high affinity D2-like ligands that have been used as PET ligands. Of the substituted benzamides, sulpiride is often considered the prototype, and has been widely studied, but only amisulpride (Figure 5) is currently used clinically although many other members of this class have shown varying degrees of clinical efficacy and side effects. Amisulpride has D2/D3 selectivity, and low affinity for other dopamine or serotonergic, alpha-adrenergic, histaminergic or cholinergic receptors. It is equieffective to haloperidol and α-flupenthixol for positive symptoms, and at lower does is reported to be significantly more effective in reducing primary negative symptoms than placebo. The clinical properties of amisulpride appear more “atypical” than other members of this family (e.g., sulpiride) despite having similar receptor profiles. The underlying mechanisms for this are unclear, but may well involve functionally selective signaling at the D2 or other receptors.
Other compounds with partial agonist/functionally selective D2-like properties
There are many hints in the literature of compounds that might have functionally selective properties at D2 receptors. Talipexole (B-HT 920) initially was suggested as a partial agonist at dopamine D2 receptors because of its ability to inhibit both synthesis and utilization of dopamine but not produce any stereotypies on hyperactivity. Studies with stimulation of presynaptic D2 dopamine receptors by talipexole in comparision with apomorphine inhibited the synthesis of dopamine in the corpus striatum of γ-butyrolactone-treated mice to same extent suggesting that B-HT 920 to be a full agonist at presynaptic D2 dopamine receptors [145]. Although tested against negative symptoms of schizophrenia [146], talipexole later became a candidate for Parkinson's disease therapy. SDZ HDC 912 is an ergoline derivative dopamine D2 partial agonist that has intrinsic agonist activity adequate to maintain dopaminergic transmission sufficient to avoid extrapyramidal side-effects as well as hyperprolactinemia [147]. Although the compound had partial agonist properties like the aripiprazole precursor OPC-4392, it also differed in several ways, suggesting it might be functionally selective [148].
Roxindole (EMD 49980) was reported to be a selective agonist of presynaptic dopamine D2 receptors with little D1 affinity. It is heuristically less interesting because it has higher affinity for the D3 and D4 receptors, and weak partial agonist activity at D2 receptors [149]. Other compounds of potential interest include UH232 [23, 150], S32504 [151], SDZ 208-912 [152, 153], PD 143188 (CI-1007) [154], SLV-308 (SME-308) [155, 156], F15063 [157, 158], SSR181507 [159], (-)-OSU6162 (PNU-96391A; Figure 5) [160, 161], ACR16 [162, 163], and many others. What is of particular importance is that there has been the view that dopamine stabilization (i.e., the unique clinical effects of aripiprazole) can be explained simply “partial agonist intrinsic activity.” Thus, Tadori et al. [164] compared aripiprazole, bifeprunox, SDZ 208-912, OPC-4392, and ACR16 in terms of degrees of intrinsic activity (i.e., inhibition of forskolinstimulated cAMP accumulation) in clonal CHO cell lines expressing high and low densities of human dopamine D2L and D2S receptors. In cells with lower receptor densities, all drugs had partial intrinsic activity (SDZ 208-912 < aripiprazole < bifeprunox=OPC-4392). They concluded that this specific degree of partial agonism was the reason for efficacy in both positive and negative symptoms of schizophrenia, as well as the demonstrated low liability for parkinsonism and hyperprolactinemia. We believe that this explanation fails to account for the clinical failure of preclamol, a compound with intrinsic activity similar to aripiprazole in many systems. Coupled with the fact that aripiprazole has varied intrinsic activity depending on the assay system [67, 69, 70], we feel that it is timely to consider the complexity of D2-mediated signaling as an alternate mechanism of importance.
b. Examples of recent pipeline drugs
Bifeprunox (DU 127090) (Figure 5) is a novel atypical antipsychotic candidate whose early development was by Solvay-Lundbeck pharmaceuticals. It is a full-spectrum dopamine D2 receptor partial agonist and also possesses 5-HT1A receptor agonism. It exhibits low intrinsic activity at the D2 receptor and high affinity and moderate to high intrinsic activity at the 5-HT1A receptor. Interpreted as a partial D2 agonist with 5-HT1A activity, it is theoretically similar to aripiprazole. Although bifeprunox was shown to be more effective than placebo in the treatment of schizophrenia [165], the US FDA rejected an NDA for it on both efficacy and safety grounds [166]. If functional selectivity rather than partial agonism is indeed the critical mechanism, this leads to the hypothesis that the signaling profile of bifeprunox and aripiprazole differ.
Aplindore (DAB-452) (Figure 5) was another of the partial agonist family that was in development at Wyeth as an atypical antipsychotic. Aplindore has high affinity for D2/D3 receptors, but unlike aripiprazole, has low affinity for 5-HT1A. In a variety of assays (GTPγS binding, MAPK, and calcium flux), the intrinsic activities of aplindore are higher than aripiprazole, and unlike aripiprazole, aplindore induced D2-dependent contralateral turning in unilaterally 6-OHDA lesioned rats [167]. The schizophrenia indication for aplindore has now been abandoned, and Neurogen is pursuing indications for Parkinson's disease and restless leg syndrome. These data may be interpreted as additional evidence for the importance of functional selectivity, or that the intrinsic activity of a partial agonist must be very low for the drug to be effective in schizophrenia.
4. Challenges for discovery of functionally selective drugs
As the foregoing material makes clear, we believe that the actions of aripiprazole are due to its functionally selective D2 dopamine receptor properties, as well as possibly involving off-site receptors such as the 5-HT1A. This hypothesis suggests that even better drugs may be possible by understanding the specific signaling mechanisms that affect the actions of aripiprazole in humans. In this regard, almost every laboratory that played a role in bringing functional selectivity into the pharmacological mainstream also concluded that this mechanism was both a great complication and a great opportunity for drug discovery [72]. As the pharmaceutical industry has become aware of both the benefits and challenges, the obvious question is how to incorporate this mechanism into the discovery of novel compounds.
The lack of a totally predictive animal model for complex psychiatric disorders like schizophrenia is a major research handicap. Indeed, the validity of our models is often dependent on the fact that they respond to current drugs, creating some inherent circular logic. This complicates the selection of novel compounds that might enter preclinical development. Functional selectivity, per se, complicates the issues inherent in setting the parameters for high- or medium throughput screening. Even accepting that aripiprazole may work primarily as a functionally selective D2 ligand (as opposed to a partial agonist), it is challenging to decide how to assay next-generation aripiprazole-like ligands to select the best of the potential drug candidates. One of the critical issues may be the host cell line to study. The properties of a drug candidate when studied at D2 receptors expressed heterologously in non-neural cell lines (e.g., HEK293 or CHO) may differ from neurally-derived lines (e.g., C6, MN9D, SK-N-MC, etc.). Of course, even responses in the latter may differ from those of native neurons. These issues are clearly illustrated in the milieu-specific effects that have been shown for aripiprazole [67, 69, 70]. This issue has critical ramifications for drug discovery, especially in the adaptation of systems that while providing very high throughput and high Z-scores, may not always reflect the relevant properties of ligands that are being screened. The impact of cellular milieu cannot be overstated.
As one recent example, Heusler et al. [158] studied the effects of antipsychotic drugs or drug candidates to cause internalization of the human D2S receptor using hemaglutinin (HA)-tags in HEK293 cells in which there was also increased co-expression of G protein-coupled receptor kinase 2 (GRK2) and β-arrestin2. Dopamine, quinpirole and bromocriptine behaved as full agonists, whereas S(-)-3-(3-hydroxyphenyl)-N-n-propylpiperidine [preclamol (-)-3PPP] and sarizotan were partial agonists. All of the antipsychotics tested (haloperidol, olanzapine, nemonapride, ziprasidone and clozapine) failed to internalize D2S receptors, whereas aripiprazole had >50% intrinsic activity. Bifeprunox and SSR181507 also were partial agonists whereas SLV313 and F15063) were inactive. Conversely, Urban et al. [72] used FLAG-tagged D2L receptors ands reported that neither preclamol nor aripiprazole had intrinsic activity. These data underscore how subtle experimental differences may lead to very different conclusions. It is unclear whether the differences were due to the nature of the receptor tag, the use of short- versus long isoform, or the co-expression of GRK2 and β-arrestin2, or some other factor. The task of extrapolating or integrating in vitro functional selectivity findings thus remains a major challenge. We hope that the examples we have provided, however, suggest that demonstrating partial agonism in a single, often non-physiological, assay system is neither mechanistically informative, nor useful in selecting novel drug candidates.
5. Is the “generational” nomenclature right for antipsychotic drugs?
For the last half-century, all approved antipsychotic drugs have had affinity for dopamine D2-like receptors as an apparently essential aspect of their mechanism of action. There is no fundamental neurobiological reason, however, that requires this to always be the case. Conversely, there is evidence that several non-dopaminergic approaches might lead to a novel antipsychotic drug. Thus, there has been interest in selective ligands for various serotonin receptors (5-HT2A, 5-HT2C, 5-HT7), whether these be agonists, antagonists, or functionally selective ligands. Another research focus has been on glutamatergic interventions, either via modulation of ionotropic or metabotropic glutamate transmission. Moreover, novel therapies may come from treatment of currently resistant facets of schizophrenia (e.g., decreasing cognitive deficits via α7 nicotinic agonists or D1 dopamine agonists), or from targets with unexpected actions (e.g., drugs acting at a previously orphan receptor, acting at a known receptor with unexpected effects, or even a more distal aspect of a signaling cascade).
In the interest of effective communication, we would suggest that the field should consider a nomenclature that will accommodate novel directions that are likely to emerge. If indeed several of the therapeutic directions noted above come to pass, the use of “X generation” terminology may quickly lead to a Tower of Babel that, while interpretable to those immersed in the field, will be incomprehensible to practicing physicians, residents, medical students, and even those scientists in closely aligned areas. In other fields (antibiotics, anticancer, etc.), drugs are often categorized according to their mechanism(s) of action. One wonders if communication about antipsychotic drugs might not benefit in a similar way. Thus we could have from the current drugs: “antipsychotics-dopamine antagonists” (1st generation); “antipsychoticsdopamine-serotonin antagonists” (some 2nd generation); “antipsychotics-multi-targeted” (other 2nd generation); and “antipsychotics-dopamine-functionally selective” (3rd generation). This would then allow growth into possible future classes: “antipsychotics-serotonin 5-HT2A functionally selective”; “antipsychotics-dopamine D1 agonists”; “antipsychotics-glutamate metabotropic allosteric modulators”; etc. Although not as simple and elegant as “X Generation antipsychotic,” it might facilitate communication among Generation X scientists.
Acknowledgments
This work was supported, in part, by Public Health Service research grants MH082441 and MH040537.
Footnotes
Conflict of interest: Richard Mailman has a potential conflict of interest as regards a financial interest in Biovalve Technologies and Effipharma Inc. who have licenses for some cited compounds or their offshoots. This conflict of interest is monitored by the Pennsylvania State University. All opinions in this manuscript are those of the authors alone, and not of Penn State University, Biovalve Technologies, or Effipharma Inc.
Reference List
- 1.Delay J, Deniker P, Harl JM. Therapeutic method derived from hiberno-therapy in excitation and agitation states. Ann Med Psychol (Paris) 1952;110:267–273. [PubMed] [Google Scholar]
- 2.Carlsson A, Lindqvist M. Effect of chlorpromazine and haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- 3.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 4.Ungerstedt U. Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl. 1971;367:1–48. doi: 10.1111/j.1365-201x.1971.tb10998.x. [DOI] [PubMed] [Google Scholar]
- 5.Kebabian JW, Petzold GL, Greengard P. Dopamine-sensitive adenylate cyclase in caudate nucleus of rat brain, and its similarity to the “dopamine receptor”. Proc Natl Acad Sci U S A. 1972;69:2145–2149. doi: 10.1073/pnas.69.8.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clement-Cormier YC, Kebabian JW, Petzold GL, Greengard P. Dopamine-sensitive adenylate cyclase in mammalian brain: a possible site of action of antipsychotic drugs. Proc Natl Acad Sci U S A. 1974;71:1113–1117. doi: 10.1073/pnas.71.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iversen LL. Dopamine receptors in the brain. Science. 1975;188:1084–1089. doi: 10.1126/science.2976. [DOI] [PubMed] [Google Scholar]
- 8.Trabucchi M, Longoni R, Fresia P, Spano PF. Sulpiride: a study of the effects on dopamine receptors in rat neostriatum and limbic forebrain. Life Sci. 1975;17:1551–1556. doi: 10.1016/0024-3205(75)90176-9. [DOI] [PubMed] [Google Scholar]
- 9.Garau L, Govoni S, Stefanini E, Trabucchi M, Spano PF. Dopamine receptors: pharmacological and anatomical evidences indicate that two distinct dopamine receptor populations are present in rat striatum. Life Sci. 1978;23:1745–1750. doi: 10.1016/0024-3205(78)90102-9. [DOI] [PubMed] [Google Scholar]
- 10.Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277:93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- 11.Neve KA, Neve RL. The dopamine receptors. Humana Press; Totowa, N.J.: 1997. [Google Scholar]
- 12.Jenner P, Demirdemar R. Dopamine receptor sub-types: from basic sciences to clinical applications. IOS Press; Burke, VA: 1997. [Google Scholar]
- 13.Sealfon SC, Olanow CW. Dopamine receptors: from structure to behavior. Trends Neurosci. 2000;23:S34–S40. doi: 10.1016/s1471-1931(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Lawler CP, Lewis MM, Nichols DE, Mailman RB. D1 dopamine receptors. Int Rev Neurobiol. 2001;48:65–139. doi: 10.1016/s0074-7742(01)48014-7. [DOI] [PubMed] [Google Scholar]
- 15.Dearry A, Gingrich JA, Falardeau P, Fremeau RT, Jr, Bates MD, Caron MG. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature. 1990;347:72–76. doi: 10.1038/347072a0. [DOI] [PubMed] [Google Scholar]
- 16.Monsma FJJ, Mahan LC, McVittie LD, Gerfen CR, Sibley DR. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci U S A. 1990;87:6723–6727. doi: 10.1073/pnas.87.17.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou QY, Grandy DK, Thambi L, Kushner JA, van Tol HH, Cone R, et al. Cloning and expression of human and rat D1 dopamine receptors. Nature. 1990;347:76–80. doi: 10.1038/347076a0. [DOI] [PubMed] [Google Scholar]
- 18.Sunahara RK, Guan HC, O'Dowd BF, Seeman P, Laurier LG, Ng G, et al. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature. 1991;350:614–619. doi: 10.1038/350614a0. [DOI] [PubMed] [Google Scholar]
- 19.Dal Toso R, Sommer B, Ewert M, Herb A, Pritchett DB, Bach A, et al. The dopamine D2 receptor: two molecular forms generated by alternative splicing. EMBO J. 1989;8:4025–4034. doi: 10.1002/j.1460-2075.1989.tb08585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giros B, Sokoloff P, Martres MP, Riou JF, Emorine LJ, Schwartz JC. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature. 1989;342:923–926. doi: 10.1038/342923a0. [DOI] [PubMed] [Google Scholar]
- 21.Monsma FJJ, McVittie LD, Gerfen CR, Mahan LC, Sibley DR. Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature. 1989;342:926–929. doi: 10.1038/342926a0. [DOI] [PubMed] [Google Scholar]
- 22.Chio CL, Hess GF, Graham RS, Huff RM. A second molecular form of D2 dopamine receptor in rat and bovine caudate nucleus. Nature. 1990;343:266–269. doi: 10.1038/343266a0. [DOI] [PubMed] [Google Scholar]
- 23.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 24.van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, et al. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature. 1991;350:610–614. doi: 10.1038/350610a0. [DOI] [PubMed] [Google Scholar]
- 25.O'Malley KL, Harmon S, Tang L, Todd RD. The rat dopamine D4 receptor: sequence, gene structure, and demonstration of expression in the cardiovascular system. New Biol. 1992;4:137–146. [PubMed] [Google Scholar]
- 26.Mill J, Curran S, Kent L, Richards S, Gould A, Virdee V, et al. Attention deficit hyperactivity disorder (ADHD) and the dopamine D4 receptor gene: evidence of association but no linkage in a UK sample. Mol Psychiatry. 2001;6:440–444. doi: 10.1038/sj.mp.4000881. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt LA, Fox NA, Perez-Edgar K, Hu S, Hamer DH. Association of DRD4 with attention problems in normal childhood development. Psychiatr Genet. 2001;11:25–29. doi: 10.1097/00041444-200103000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Swanson JM, Sunohara GA, Kennedy JL, Regino R, Fineberg E, Wigal T, et al. Association of the dopamine receptor D4 (DRD4) gene with a refined phenotype of attention deficit hyperactivity disorder (ADHD): a family-based approach. Mol Psychiatry. 1998;3:38–41. doi: 10.1038/sj.mp.4000354. [DOI] [PubMed] [Google Scholar]
- 29.Jonsson EG, Ivo R, Gustavsson JP, Geijer T, Forslund K, Mattila-Evenden M, et al. No association between dopamine D4 receptor gene variants and novelty seeking. Mol Psychiatry. 2002;7:18–20. doi: 10.1038/sj.mp.4000950. [DOI] [PubMed] [Google Scholar]
- 30.Jonsson EG, Ivo R, Forslund K, Mattila-Evenden M, Rylander G, Cichon S, et al. No association between a promoter dopamine D(4) receptor gene variant and schizophrenia. Am J Med Genet. 2001;105:525–528. doi: 10.1002/ajmg.1478. [DOI] [PubMed] [Google Scholar]
- 31.Kotler M, Manor I, Sever Y, Eisenberg J, Cohen H, Ebstein RP, et al. Failure to replicate an excess of the long dopamine D4 exon III repeat polymorphism in ADHD in a family-based study. Am J Med Genet. 2000;96:278–281. doi: 10.1002/1096-8628(20000612)96:3<278::aid-ajmg8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 32.Kane JM. The current status of neuroleptic therapy. J Clinical Psychiatry. 1989;50:322–328. [PubMed] [Google Scholar]
- 33.Fleischhacker WW. New developments in the pharmacotherapy of schizophrenia. J Neural Transm Suppl. 2003:105–117. doi: 10.1007/978-3-7091-6020-6_7. [DOI] [PubMed] [Google Scholar]
- 34.Barnes TR, McPhillips MA. Critical analysis and comparison of the side-effect and safety profiles of the new antipsychotics. Br J Psychiatry Suppl. 1999:34–43. [PubMed] [Google Scholar]
- 35.Kane JM. Pharmacologic treatment of schizophrenia. Biol Psychiatry. 1999;46:1396–1408. doi: 10.1016/s0006-3223(99)00059-1. [DOI] [PubMed] [Google Scholar]
- 36.Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10:79–104. doi: 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto S, Duncan GE, Mailman RB, Lieberman JA. Developing novel antipsychotic drugs: strategies and goals. Curr Opin CPNS Invest Drugs. 2000;2:25–39. [Google Scholar]
- 38.Kane J. Olanzapine in the long-term treatment of schizophrenia. Br J Psychiatry Suppl. 1999:26–29. [PubMed] [Google Scholar]
- 39.Lieberman JA. Atypical antipsychotic drugs as a first-line treatment of schizophrenia: a rationale and hypothesis. J Clin Psychiatry. 1996;57 11:68–71. [PubMed] [Google Scholar]
- 40.Meyer JM, Simpson GM. From chlorpromazine to olanzapine: a brief history of antipsychotics. Psychiatr Serv. 1997;48:1137–1139. doi: 10.1176/ps.48.9.1137. [DOI] [PubMed] [Google Scholar]
- 41.Niedzwiecki DM, Mailman RB, Cubeddu LX. Greater potency of mesoridazine and sulforidazine compared with the parent compound, thioridazine, on striatal dopamine autoreceptors. J Pharmacol Exp Ther. 1984;228:636–639. [PubMed] [Google Scholar]
- 42.Niedzwiecki DM, Cubeddu LX, Mailman RB. Comparative anticholinergic properties of thioridazine, mesoridazine and sulforidazine. J Pharmacol Exp Ther. 1989;250:126–133. [PubMed] [Google Scholar]
- 43.Niedzwiecki DM, Cubeddu LX, Mailman RB. Comparative antidopaminergic properties of thioridazine, mesoridazine and sulforidazine on the corpus striatum. J Pharmacol Exp Ther. 1989;250:117–125. [PubMed] [Google Scholar]
- 44.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 45.Lieberman J, Johns C, Cooper T, Pollack S, Kane J. Clozapine pharmacology and tardive dyskinesia. Psychopharmacology. 1989;99(Suppl):S54–S59. doi: 10.1007/BF00442560. [DOI] [PubMed] [Google Scholar]
- 46.Lieberman JA. Understanding the mechanism of action of atypical antipsychotic drugs. A review of compounds in use and development. Br J Psychiatry Suppl. 1993:7–18. [PubMed] [Google Scholar]
- 47.Bymaster FP, Calligaro DO, Falcone JF, Marsh RD, Moore NA, Tye NC, et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology. 1996;14:87–96. doi: 10.1016/0893-133X(94)00129-N. [DOI] [PubMed] [Google Scholar]
- 48.Brown CS, Markowitz JS, Moore TR, Parker NG. Atypical antipsychotics: Part II: Adverse effects, drug interactions, and costs. Ann Pharmacother. 1999;33:210–217. doi: 10.1345/aph.17216. [DOI] [PubMed] [Google Scholar]
- 49.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively nonselective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 50.Stroup TS, Lieberman JA, McEvoy JP, Davis SM, Swartz MS, Keefe RS, et al. Results of phase 3 of the CATIE schizophrenia trial. Schizophr Res. 2009;107:1–12. doi: 10.1016/j.schres.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huttunen M. The evolution of the Serotonin-Dopamine Antagonist Concept. J Clin Psychopharmacol. 1995;15:4S–10S. doi: 10.1097/00004714-199502001-00002. [DOI] [PubMed] [Google Scholar]
- 52.Meltzer HY, Matsubara S, Lee JC. The ratios of serotonin2 and dopamine2 affinities differentiate atypical and typical antipsychotic drugs. Psychopharmacol Bull. 1989;25:390–392. [PubMed] [Google Scholar]
- 53.Markowitz JS, Brown CS, Moore TR. Atypical antipsychotics. Part I: Pharmacology, pharmacokinetics, and efficacy. Ann Pharmacother. 1999;33:73–85. doi: 10.1345/aph.17215. [DOI] [PubMed] [Google Scholar]
- 54.Chouinard G. Effects of risperidone in tardive dyskinesia: an analysis of the Canadian multicenter risperidone study. J Clin Psychopharmacol. 1995;15:36S–44S. doi: 10.1097/00004714-199502001-00007. [DOI] [PubMed] [Google Scholar]
- 55.Feenstra MG, Sumners C, Goedemoed JH, de Vries JB, Rollema H, Horn AS. A comparison of the potencies of various dopamine receptor agonists in models for pre- and postsynaptic receptor activity. Naunyn Schmiedebergs Arch Pharmacol. 1983;324:108–115. doi: 10.1007/BF00497015. [DOI] [PubMed] [Google Scholar]
- 56.Meller E, Bohmaker K, Namba Y, Friedhoff AJ, Goldstein M. Relationship between receptor occupancy and response at striatal dopamine autoreceptors. Mol Pharmacol. 1987;31:592–598. [PubMed] [Google Scholar]
- 57.Kenakin TP. Pharmacologic analysis of drug-receptor interaction. Lippincott-Raven Publishers; Philadelphia: 1997. [Google Scholar]
- 58.Tamminga CA, Schaffer MH, Smith RC, Davis JM. Schizophrenic symptoms improve with apomorphine. Science. 1978;200:567–568. doi: 10.1126/science.347574. [DOI] [PubMed] [Google Scholar]
- 59.Corsini GU, Pitzalis GF, Bernardi F, Bocchetta A, Del ZM. The use of dopamine agonists in the treatment of schizophrenia. Neuropharmacology. 1981;20:1309–1313. [PubMed] [Google Scholar]
- 60.Smith RC, Tamminga C, Davis JM. Effect of apomorphine on schizophrenic symptoms. J Neural Transm. 1977;40:171–176. doi: 10.1007/BF01250567. [DOI] [PubMed] [Google Scholar]
- 61.Tamminga CA, Gotts MD, Thaker GK, Alphs LD, Foster NL. Dopamine agonist treatment of schizophrenia with N-propylnorapomorphine. Arch Gen Psychiatry. 1986;43:398–402. doi: 10.1001/archpsyc.1986.01800040108015. [DOI] [PubMed] [Google Scholar]
- 62.Lahti AC, Weiler MA, Corey PK, Lahti RA, Carlsson A, Tamminga CA. Antipsychotic properties of the partial dopamine agonist (-)-3-(3-hydroxyphenyl)-N-n-propylpiperidine(preclamol) in schizophrenia. Biol Psychiatry. 1998;43:2–11. doi: 10.1016/s0006-3223(97)00030-9. [DOI] [PubMed] [Google Scholar]
- 63.Tamminga CA, Cascella NG, Lahti RA, Lindberg M, Carlsson A. Pharmacologic properties of (-)-3PPP (preclamol) in man. J Neural Transm Gen Sect. 1992;88:165–175. doi: 10.1007/BF01244730. [DOI] [PubMed] [Google Scholar]
- 64.Tamminga CA. Partial dopamine agonists in the treatment of psychosis. J Neural Transm. 2002;109:411–420. doi: 10.1007/s007020200033. [DOI] [PubMed] [Google Scholar]
- 65.Stahl SM. Dopamine system stabilizers, aripiprazole, and the next generation of antipsychotics, part 1, “Goldilocks” actions at dopamine receptors. J Clin Psychiatry. 2001;62:841–842. doi: 10.4088/jcp.v62n1101. [DOI] [PubMed] [Google Scholar]
- 66.Lieberman JA. Dopamine partial agonists: a new class of antipsychotic. CNS Drugs. 2004;18:251–267. doi: 10.2165/00023210-200418040-00005. [DOI] [PubMed] [Google Scholar]
- 67.Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA, et al. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology. 1999;20:612–627. doi: 10.1016/S0893-133X(98)00099-2. [DOI] [PubMed] [Google Scholar]
- 68.Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther. 2002;302:381–389. doi: 10.1124/jpet.102.033175. [DOI] [PubMed] [Google Scholar]
- 69.Shapiro DA, Renock S, Arrington E, Sibley DR, Chiodo LA, Roth BL, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 70.Urban JD, Vargas GA, von Zastrow M, Mailman RB. Aripiprazole has Functionally Selective Actions at Dopamine D(2) Receptor-Mediated Signaling Pathways. Neuropsychopharmacology. 2007;32:67–77. doi: 10.1038/sj.npp.1301071. [DOI] [PubMed] [Google Scholar]
- 71.Kikuchi T, Tottori K, Uwahodo Y, Hirose T, Miwa T, Oshiro Y, et al. 7-(4-[4-(2,3-Dichlorophenyl)-1-piperazinyl]butyloxy)-3,4-dihydro-2(1H)-qui nolinone (OPC-14597), a new putative antipsychotic drug with both presynaptic dopamine autoreceptor agonistic activity and postsynaptic D2 receptor antagonistic activity. J Pharmacol Exp Ther. 1995;274:329–336. [PubMed] [Google Scholar]
- 72.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 73.Stephenson RP. A modification of receptor theory. Br J Pharmacol. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kenakin T. Pharmacologic analysis of drug-receptor interaction. Lippincott-Raven; Philadelphia: 1997. [Google Scholar]
- 75.Furchgott RF. The use of -haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. In: Harper NJ, Simmonds AB, editors. Advances in Drug Research. Academic Press; New York: 1966. pp. 21–55. [Google Scholar]
- 76.Lovenberg TW, Brewster WK, Mottola DM, Lee RC, Riggs RM, Nichols DE, et al. Dihydrexidine, a novel selective high potency full dopamine D-1 receptor agonist. Eur J Pharmacol. 1989;166:111–113. doi: 10.1016/0014-2999(89)90690-0. [DOI] [PubMed] [Google Scholar]
- 77.Brewster WK, Nichols DE, Riggs RM, Mottola DM, Lovenberg TW, Lewis MH, et al. trans-10,11-dihydroxy-5,6,6a,7,8,12b-hexahydrobenzo[a]phenanthridine: a highly potent selective dopamine D1 full agonist. J Med Chem. 1990;33:1756–1764. doi: 10.1021/jm00168a034. [DOI] [PubMed] [Google Scholar]
- 78.Mottola DM, Brewster WK, Cook LL, Nichols DE, Mailman RB. Dihydrexidine, a novel full efficacy D1 dopamine receptor agonist. J Pharmacol Exp Ther. 1992;262:383–393. [PubMed] [Google Scholar]
- 79.Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, et al. Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther. 2002;301:1166–1178. doi: 10.1124/jpet.301.3.1166. [DOI] [PubMed] [Google Scholar]
- 80.Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS, et al. Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J Pharmacol Exp Ther. 2002;301:1179–1189. doi: 10.1124/jpet.301.3.1179. [DOI] [PubMed] [Google Scholar]
- 81.Mailman RB, Nichols DE, Lewis MM, Blake BL, Lawler CP. Functional Effects of Novel Dopamine Ligands: Dihydrexidine and Parkinson's Disease as a First Step. In: Jenner P, Demirdemar R, editors. Dopamine Receptor Subtypes: From Basic Science to Clinical Application. IOS Stockton Press; 1998. pp. 64–83. [Google Scholar]
- 82.Roth BL, Chuang DM. Multiple mechanisms of serotonergic signal transduction. Life Sci. 1987;41:1051–1064. doi: 10.1016/0024-3205(87)90621-7. [DOI] [PubMed] [Google Scholar]
- 83.Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- 84.Darney KJ, Jr, Lewis MH, Brewster WK, Nichols DE, Mailman RB. Behavioral effects in the rat of dihydrexidine, a high-potency, full-efficacy D1 dopamine receptor agonist. Neuropsychopharmacology. 1991;5:187–195. [PubMed] [Google Scholar]
- 85.Eden RJ, Costall B, Domeney AM, Gerrard PA, Harvey CA, Kelly ME, et al. Preclinical pharmacology of ropinirole (SK&F 101468-A) a novel dopamine D2 agonist. Pharmacol Biochem Behav. 1991;38:147–154. doi: 10.1016/0091-3057(91)90603-y. [DOI] [PubMed] [Google Scholar]
- 86.Smith HP, Nichols DE, Mailman RB, Lawler CP. Locomotor inhibition, yawning and vacuous chewing induced by a novel dopamine D2 post-synaptic receptor agonist. Eur J Pharmacol. 1997;323:27–36. doi: 10.1016/s0014-2999(97)00026-5. [DOI] [PubMed] [Google Scholar]
- 87.Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy- dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24:485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- 88.Friedman JH, Berman RM, Goetz CG, Factor SA, Ondo WG, Wojcieszek J, et al. Open-label flexible-dose pilot study to evaluate the safety and tolerability of aripiprazole in patients with psychosis associated with Parkinson's disease. Mov Disord. 2006;21:2078–2081. doi: 10.1002/mds.21091. [DOI] [PubMed] [Google Scholar]
- 89.Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- 90.Mailman RB, Wang YM, Kant A, Brown JT. Functional selectivity of dopamine receptors. In: Neve KA, editor. Functional selectivity of G protein-coupled receptor ligands. Humana; New York: 2009. pp. 177–209. [Google Scholar]
- 91.De Camilli P, Macconi D, Spada A. Dopamine inhibits adenylate cyclase in human prolactin-secreting pituitary adenomas. Nature. 1979;278:252–254. doi: 10.1038/278252a0. [DOI] [PubMed] [Google Scholar]
- 92.Stoof JC, Kebabian JW. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–368. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- 93.Huff RM. Signaling pathways modulated by dopamine receptors. In: Neve KA, Neve RL, editors. The Dopamine Receptors. Humana Press; Totowa, NJ: 1997. pp. 167–192. [Google Scholar]
- 94.Huff RM, Chio CL, Lajiness ME, Goodman LV. Signal transduction pathways modulated by D2-like dopamine receptors. Adv Pharmacol. 1998;42:454–457. doi: 10.1016/s1054-3589(08)60786-3. [DOI] [PubMed] [Google Scholar]
- 95.Oxford GS, Wagoner PK. The inactivating K+ current in GH3 pituitary cells and its modification by chemical reagents. J Physiol (Lond) 1989;410:587–612. doi: 10.1113/jphysiol.1989.sp017550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol (Lond) 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu L, Shen RY, Kapatos G, Chiodo LA. Dopamine neuron membrane physiology: characterization of the transient outward current (IA) and demonstration of a common signal transduction pathway for IA and IK. Synapse. 1994;17:230–240. doi: 10.1002/syn.890170404. [DOI] [PubMed] [Google Scholar]
- 98.Greif GJ, Lin YJ, Liu JC, Freedman JE. Dopamine-modulated potassium channels on rat striatal neurons: specific activation and cellular expression. J Neurosci. 1995;15:4533–4544. doi: 10.1523/JNEUROSCI.15-06-04533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cass WA, Zahniser NR. Potassium channel blockers inhibit D2 dopamine, but not A1 adenosine, receptor-mediated inhibition of striatal dopamine release. J Neurochem. 1991;57:147–152. doi: 10.1111/j.1471-4159.1991.tb02109.x. [DOI] [PubMed] [Google Scholar]
- 100.Memo M, Missale C, Carruba MO, Spano PF. D2 dopamine receptors associated with inhibition of dopamine release from rat neostriatum are independent of cyclic AMP. Neurosci Lett. 1986;71:192–196. doi: 10.1016/0304-3940(86)90557-4. [DOI] [PubMed] [Google Scholar]
- 101.Davila V, Yan Z, Craciun LC, Logothetis D, Sulzer D. D3 dopamine autoreceptors do not activate G-protein-gated inwardly rectifying potassium channel currents in substantia nigra dopamine neurons. J Neurosci. 2003;23:5693–5697. doi: 10.1523/JNEUROSCI.23-13-05693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lledo PM, Homburger V, Bockaert J, Vincent JD. Differential G protein-mediated coupling of D2 dopamine receptors to K+ and Ca2+ currents in rat anterior pituitary cells. Neuron. 1992;8:455–463. doi: 10.1016/0896-6273(92)90273-g. [DOI] [PubMed] [Google Scholar]
- 103.Seabrook GR, Knowles M, Brown N, Myers J, Sinclair H, Patel S, et al. Pharmacology of high-threshold calcium currents in GH4C1 pituitary cells and their regulation by activation of human D2 and D4 dopamine receptors. Br J Pharmacol. 1994;112:728–734. doi: 10.1111/j.1476-5381.1994.tb13138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seabrook GR, Mcallister G, Knowles MR, Myers J, Sinclair H, Patel S, et al. Depression of high-threshold calcium currents by activation of human D2 (short) dopamine receptors expressed in differentiated NG108-15 cells. Br J Pharmacol. 1994;111:1061–1066. doi: 10.1111/j.1476-5381.1994.tb14852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuzhikandathil EV, Oxford GS. Activation of human D3 dopamine receptor inhibits P/Q-type calcium channels and secretory activity in AtT-20 cells. J Neurosci. 1999;19:1698–1707. doi: 10.1523/JNEUROSCI.19-05-01698.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okada Y, Miyamoto T, Toda K. Dopamine modulates a voltage-gated calcium channel in rat olfactory receptor neurons. Brain Res. 2003;968:248–255. doi: 10.1016/s0006-8993(03)02267-4. [DOI] [PubMed] [Google Scholar]
- 107.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci U S A. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. Prog Brain Res. 1993;99:309–324. doi: 10.1016/s0079-6123(08)61354-0. [DOI] [PubMed] [Google Scholar]
- 109.Voyno-Yasenetskaya T, Conklin BR, Gilbert RL, Hooley R, Bourne HR, Barber DL. G alpha 13 stimulates Na-H exchange. J Biol Chem. 1994;269:4721–4724. [PubMed] [Google Scholar]
- 110.Huff RM. Signal transduction pathways modulated by the D2 subfamily of dopamine receptors. Cell Signal. 1996;8:453–459. doi: 10.1016/s0898-6568(96)00074-5. [DOI] [PubMed] [Google Scholar]
- 111.Luo Y, Kokkonen GC, Wang X, Neve KA, Roth GS. D2 dopamine receptors stimulate mitogenesis through pertussis toxin-sensitive G proteins and Ras-involved ERK and SAP/JNK pathways in rat C6-D2L glioma cells. J Neurochem. 1998;71:980–990. doi: 10.1046/j.1471-4159.1998.71030980.x. [DOI] [PubMed] [Google Scholar]
- 112.Welsh GI, Hall DA, Warnes A, Strange PG, Proud CG. Activation of microtubule-associated protein kinase (Erk) and p70 S6 kinase by D2 dopamine receptors. J Neurochem. 1998;70:2139–2146. doi: 10.1046/j.1471-4159.1998.70052139.x. [DOI] [PubMed] [Google Scholar]
- 113.Choi EY, Jeong D, Won K, Park, Baik JH. G protein-mediated mitogen-activated protein kinase activation by two dopamine D2 receptors. Biochem Biophys Res Commun. 1999;256:33–40. doi: 10.1006/bbrc.1999.0286. [DOI] [PubMed] [Google Scholar]
- 114.Ghahremani MH, Forget C, Albert PR. Distinct roles for Galphai2 and Gbetagamma in signaling to DNA synthesis and Galpha(i)3 in cellular transformation by dopamine D2S receptor activation in BALB/c 3T3 cells. Mol Cell Biol. 2000;20:1497–1506. doi: 10.1128/mcb.20.5.1497-1506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oak JN, Lavine N, van Tol HH. Dopamine D4 and D2L Receptor Stimulation of the Mitogen-Activated Protein Kinase Pathway Is Dependent on trans-Activation of the Platelet-Derived Growth Factor Receptor. Mol Pharmacol. 2001;60:92–103. doi: 10.1124/mol.60.1.92. [DOI] [PubMed] [Google Scholar]
- 116.Kim SJ, Kim MY, Lee EJ, Ahn YS, Baik JH. Distinct regulation of internalization and mitogen-activated protein kinase activation by two isoforms of the dopamine D2 receptor. Mol Endocrinol. 2004;18:640–652. doi: 10.1210/me.2003-0066. [DOI] [PubMed] [Google Scholar]
- 117.Kanterman RY, Mahan LC, Briley EM, Monsma FJ, Jr, Sibley DR, Axelrod J, et al. Transfected D2 dopamine receptors mediate the potentiation of arachidonic acid release in Chinese hamster ovary cells. Mol Pharmacol. 1991;39:364–369. [PubMed] [Google Scholar]
- 118.Piomelli D, Pilon C, Giros B, Sokoloff P, Martres MP, Schwartz JC. Dopamine activation of the arachidonic acid cascade as a basis for D1/D2 receptor synergism. Nature. 1991;353:164–167. doi: 10.1038/353164a0. [DOI] [PubMed] [Google Scholar]
- 119.Chio CL, Drong RF, Riley DT, Gill GS, Slightom JL, Huff RM. D4 dopamine receptor-mediated signaling events determined in transfected Chinese hamster ovary cells. J Biol Chem. 1994;269:11813–11819. [PubMed] [Google Scholar]
- 120.Vial D, Piomelli D. Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J Neurochem. 1995;64:2765–2772. doi: 10.1046/j.1471-4159.1995.64062765.x. [DOI] [PubMed] [Google Scholar]
- 121.Nilsson CL, Hellstrand M, Ekman A, Eriksson E. Both dopamine and the putative dopamine D3 receptor antagonist PNU-99194A induce a biphasic inhibition of phorbol ester-stimulated arachidonic acid release from CHO cells transfected with the dopamine D3 receptor. Life Sci. 1999;64:939–951. doi: 10.1016/s0024-3205(99)00020-x. [DOI] [PubMed] [Google Scholar]
- 122.Piomelli D, Greengard P. Lipoxygenase metabolites of arachidonic acid in neuronal transmembrane signalling. Trends Pharmacol Sci. 1990;11:367–373. doi: 10.1016/0165-6147(90)90182-8. [DOI] [PubMed] [Google Scholar]
- 123.DiMarzo V, Piomelli D. Participation of prostaglandin E2 in dopamine D2 receptor-dependent potentiation of arachidonic acid release. J Neurochem. 1992;59:379–382. doi: 10.1111/j.1471-4159.1992.tb08915.x. [DOI] [PubMed] [Google Scholar]
- 124.L'hirondel M, Cheramy A, Godeheu G, Glowinski J. Effects of arachidonic acid on dopamine synthesis, spontaneous release, and uptake in striatal synaptosomes from the rat. J Neurochem. 1995;64:1406–1409. doi: 10.1046/j.1471-4159.1995.64031406.x. [DOI] [PubMed] [Google Scholar]
- 125.Zhang L, Reith ME. Regulation of the functional activity of the human dopamine transporter by the arachidonic acid pathway. Eur J Pharmacol. 1996;315:345–354. doi: 10.1016/s0014-2999(96)00646-2. [DOI] [PubMed] [Google Scholar]
- 126.Mitchell R, McCulloch D, Lutz E, Johnson M, MacKenzie C, Fennell M, et al. Rhodopsin-family receptors associate with small G proteins to activate phospholipase D. Nature. 1998;392:411–414. doi: 10.1038/32937. [DOI] [PubMed] [Google Scholar]
- 127.Senogles SE. The D2s dopamine receptor stimulates phospholipase D activity: A novel signaling pathway for dopamine. Mol Pharmacol. 2000;58:455–462. doi: 10.1124/mol.58.2.455. [DOI] [PubMed] [Google Scholar]
- 128.Kurose H, Katada T, Amano T, Ui M. Specific uncoupling by islet-activating protein, pertussis toxin, of negative signal transduction via alpha-adrenergic, cholinergic, and opiate receptors in neuroblastoma x glioma hybrid cells. J Biol Chem. 1983;258:4870–4875. [PubMed] [Google Scholar]
- 129.Bokoch GM, Katada T, Northup JK, Hewlett EL, Gilman AG. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J Biol Chem. 1983;258:2072–2075. [PubMed] [Google Scholar]
- 130.Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, et al. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. 2007;27:881–885. doi: 10.1523/JNEUROSCI.5074-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, et al. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- 132.Frebel K, Wiese S. Signalling molecules essential for neuronal survival and differentiation. Biochem Soc Trans. 2006;34:1287–1290. doi: 10.1042/BST0341287. [DOI] [PubMed] [Google Scholar]
- 133.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 135.Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 136.Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- 137.Beaulieu JM, Caron MG. Beta-arrestin goes nuclear. Cell. 2005;123:755–757. doi: 10.1016/j.cell.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 138.Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, et al. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 139.Mailman RB, Huang X. Dopamine receptor pharmacology. In: Koller W, Melamed E, editors. Handbook of Clinical Neurology (3rd Series) Parkinson's Disease and Related Disorders. Elsevier; Amsterdam: 2007. pp. 77–105. [DOI] [PubMed] [Google Scholar]
- 140.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hjorth S, Clark D, Svensson K, Carlsson A, Thorberg O. Sub-chronic administration of (-)-3-PPP and central dopamine receptor sensitivity changes. J Neural Transm. 1985;64:187–198. doi: 10.1007/BF01256466. [DOI] [PubMed] [Google Scholar]
- 142.KiKuchi K, Hirata Y, Minami M, Nagatsu T. Effect of 7-(3-[4-(2,3-dimethylphenyl)piperazinyl]propoxy)- 2(1H)-quinolinone (OPC-4392), a newly synthesized agonist for presynaptic dopamine D2 receptor, on tyrosine hydroxylation in rat striatal slices. Life Sci. 1988;42:343–349. doi: 10.1016/0024-3205(88)90644-3. [DOI] [PubMed] [Google Scholar]
- 143.Yasuda Y, Kikuchi T, Suzuki S, Tsutsui M, Yamada K, Hiyama T. 7-[3-(4-[2,3-Dimethylphenyl]piperazinyl)propoxy]-2(1H)-quinolinone (OPC-4392), a presynaptic dopamine autoreceptor agonist and postsynaptic D2 receptor antagonist. Life Sci. 1988;42:1941–1954. doi: 10.1016/0024-3205(88)90493-6. [DOI] [PubMed] [Google Scholar]
- 144.Zhang X, Nakata Y, Kikuchi T, Segawa T. Interactions of 7-[3-(4-[2,3-dimethylphenyl]piperazinyl)- propoxy]-2(1H)-quinolinone binding in rat striatum: effects of lesions. Pharmaceutical Research. 1990;7:280–282. doi: 10.1023/a:1015882314302. [DOI] [PubMed] [Google Scholar]
- 145.Hinzen D, Hornykiewicz O, Kobinger W, Pichler L, Pifl C, Schingnitz G. The dopamine autoreceptor agonist B-HT 920 stimulates denervated postsynaptic brain dopamine receptors in rodent and primate models of Parkinson's disease: a novel approach to treatment. Eur J Pharmacol. 1986;131:75–86. doi: 10.1016/0014-2999(86)90517-0. [DOI] [PubMed] [Google Scholar]
- 146.Ohmori T, Koyama T, Inoue T, Matsubara S, Yamashita I. B-HT 920, a dopamine D2 agonist, in the treatment of negative symptoms of chronic schizophrenia. Biol Psychiatry. 1993;33:687–693. doi: 10.1016/0006-3223(93)90117-v. [DOI] [PubMed] [Google Scholar]
- 147.Coward D, Dixon K, Enz A, Shearman G, Urwyler S, White T, et al. Partial brain dopamine D2 receptor agonists in the treatment of schizophrenia. Psychopharmacol Bull. 1989;25:393–397. [PubMed] [Google Scholar]
- 148.Duval F, Mokrani MC, Macher JP, Crocq MA, Castro JO, Bailey P, et al. Neuroendocrine profile of SDZ HDC-912 and OPC-4392, two new atypical antipsychotic drugs, in schizophrenic patients. Psychopharmacology. 1993;110:177–180. doi: 10.1007/BF02246969. [DOI] [PubMed] [Google Scholar]
- 149.Newman-Tancredi A, Cussac D, Audinot V, Millan MJ. Actions of roxindole at recombinant human dopamine D2, D3 and D4 and serotonin 5-HT1A, 5-HT1B and 5-HT1D receptors. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:447–453. doi: 10.1007/pl00005374. [DOI] [PubMed] [Google Scholar]
- 150.Levesque D, Diaz J, Pilon C, Martres MP, Giros B, Souil E, et al. Identification, characterization, and localization of the dopamine D3 receptor in rat brain using 7-[3H]hydroxy-N,N-di-n-propyl-2- aminotetralin. Proc Natl Acad Sci U S A. 1992;89:8155–8159. doi: 10.1073/pnas.89.17.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Millan MJ, Di CB, Hill M, Jackson M, Joyce JN, Brotchie J, et al. S32504, a novel naphtoxazine agonist at dopamine D3/D2 receptors: II. Actions in rodent, primate, and cellular models of antiparkinsonian activity in comparison to ropinirole. J Pharmacol Exp Ther. 2004;309:921–935. doi: 10.1124/jpet.103.062414. [DOI] [PubMed] [Google Scholar]
- 152.Coward DM, Dixon AK, Urwyler S, Vigouret JM. Pharmacological properties of SDZ 208-912: a potential high potency, non-classical neuroleptic. Pharmacopsychiatry. 1988;21:312–313. doi: 10.1055/s-2007-1016990. [DOI] [PubMed] [Google Scholar]
- 153.Coward DM, Dixon AK, Urwyler S, White TG, Enz A, Karobath M, et al. Partial dopamine-agonistic and atypical neuroleptic properties of the amino-ergolines SDZ 208-911 and SDZ 208-912. J Pharmacol Exp Ther. 1990;252:279–285. [PubMed] [Google Scholar]
- 154.Wright JL, Caprathe BW, Downing DM, Glase SA, Heffner TG, Jaen JC, et al. The discovery and structure-activity relationships of 1,2,3,6-tetrahydro-4-phenyl-1-[(arylcyclohexenyl)alkyl]pyridines. Dopamine autoreceptor agonists and potential antipsychotic agents. J Med Chem. 1994;37:3523–3533. doi: 10.1021/jm00047a010. [DOI] [PubMed] [Google Scholar]
- 155.Glennon JC, Van SG, Ronken E, Hesselink MB, Reinders JH, Van Der Neut M, et al. In vitro characterization of SLV308 (7-[4-methyl-1-piperazinyl]-2(3H)-benzoxazolone, monohydrochloride): a novel partial dopamine D2 and D3 receptor agonist and serotonin 5-HT1A receptor agonist. Synapse. 2006;60:599–608. doi: 10.1002/syn.20330. [DOI] [PubMed] [Google Scholar]
- 156.Wolf WA. SLV-308. Solvay. Curr Opin Investig Drugs. 2003;4:878–882. [PubMed] [Google Scholar]
- 157.Heusler P, Newman-Tancredi A, Castro-Fernandez A, Cussac D. Differential agonist and inverse agonist profile of antipsychotics at D(2L) receptors coupled to GIRK potassium channels. Neuropharmacology. 2007;52:1106–1113. doi: 10.1016/j.neuropharm.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 158.Heusler P, Newman-Tancredi A, Loock T, Cussac D. Antipsychotics differ in their ability to internalise human dopamine D2S and human serotonin 5-HT1A receptors in HEK293 cells. Eur J Pharmacol. 2008;581:37–46. doi: 10.1016/j.ejphar.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 159.Claustre Y, Peretti DD, Brun P, Gueudet C, Allouard N, Alonso R, et al. SSR181507, a dopamine D(2) receptor antagonist and 5-HT(1A) receptor agonist. I: Neurochemical and electrophysiological profile. Neuropsychopharmacology. 2003;28:2064–2076. doi: 10.1038/sj.npp.1300262. [DOI] [PubMed] [Google Scholar]
- 160.Waters N, Lofberg L, Haadsma-Svensson S, Svensson K, Sonesson C, Carlsson A. Differential effects of dopamine D2 and D3 receptor antagonists in regard to dopamine release, in vivo receptor displacement and behaviour. Journal of Neural Transmission - General Section. 1994;98:39–55. doi: 10.1007/BF01277593. [DOI] [PubMed] [Google Scholar]
- 161.Tedroff J, Torstenson R, Hartvig P, Sonesson C, Waters N, Carlsson A, et al. Effects of the substituted (S)-3-phenylpiperidine (-)-OSU6162 on PET measurements in subhuman primates: evidence for tone-dependent normalization of striatal dopaminergic activity. Synapse. 1998;28:280–287. doi: 10.1002/(SICI)1098-2396(199804)28:4<280::AID-SYN3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 162.Nilsson M, Carlsson A, Markinhuhta KR, Sonesson C, Pettersson F, Gullme M, et al. The dopaminergic stabiliser ACR16 counteracts the behavioural primitivization induced by the NMDA receptor antagonist MK-801 in mice: implications for cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:677–685. doi: 10.1016/j.pnpbp.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 163.Natesan S, Svensson KA, Reckless GE, Nobrega JN, Barlow KB, Johansson AM, et al. The dopamine stabilizers (S)-(-)-(3-methanesulfonyl-phenyl)-1-propyl-piperidine [(-)-OSU6162] and 4-(3-methanesulfonylphenyl)-1-propyl-piperidine (ACR16) show high in vivo D2 receptor occupancy, antipsychotic-like efficacy, and low potential for motor side effects in the rat. J Pharmacol Exp Ther. 2006;318:810–818. doi: 10.1124/jpet.106.102905. [DOI] [PubMed] [Google Scholar]
- 164.Tadori Y, Kitagawa H, Forbes RA, McQuade RD, Stark A, Kikuchi T. Differences in agonist/antagonist properties at human dopamine D(2) receptors between aripiprazole, bifeprunox and SDZ 208-912. Eur J Pharmacol. 2007;574:103–111. doi: 10.1016/j.ejphar.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 165.Casey DE, Sands EE, Heisterberg J, Yang HM. Efficacy and safety of bifeprunox in patients with an acute exacerbation of schizophrenia: results from a randomized, double-blind, placebo-controlled, multicenter, dose-finding study. Psychopharmacology (Berl) 2008;200:317–331. doi: 10.1007/s00213-008-1207-7. [DOI] [PubMed] [Google Scholar]
- 166.Anonymous. Bifeprunox - Atypical Antipsychotic Drug. 2009 Drugdevelopment-technology.com.
- 167.Heinrich JN, Brennan J, Lai MH, Sullivan K, Hornby G, Popiolek M, et al. Aplindore (DAB-452), a high affinity selective dopamine D2 receptor partial agonist. Eur J Pharmacol. 2006;552:36–45. doi: 10.1016/j.ejphar.2006.08.063. [DOI] [PubMed] [Google Scholar]