

Summary
The switch of B cells expressing membrane-bound Igs, which serve as antigen receptors, to antibody-secreting plasmablasts and finally to non-dividing, long-lived plasma cells (PCs) lacking an antigen receptor, marks the terminal differentiation of a B cell. Antibody-secreting PCs represent the key cell type for the maintenance of a proactive humoral immunological memory. Although some populations of long-lived PCs persist in the spleen, most of them return to their ‘place of birth’ and travel to the bone marrow or invade inflamed tissues, where they survive up to several months in survival niches as resident, immobile cells. Existing data strongly support the notion that isotype-specific receptor signalling influences the migration behaviour of plasmablasts to the bone marrow. The recent observation in the murine sytem that the immigration of plasmablasts and the final differentiation to long-lived PCs in the bone marrow is dependent on the expressed B-cell isotype and the related expression of chemokine receptors leads to the conclusion that during a T-helper type 2 (Th2)-mediated immune response in wild type mice, IgE plasmablasts do not have the same chance to contribute to long-lived PC memory as IgG1 plasmablasts. The overall limited humoral IgE memory additionally restricts the quantity of IgE Igs in the serum.
Introduction
B lymphocytes derive from haematopoietic stem cells by a complex set of differentiation events. This process occurs in the foetal liver and, in adult life, in the bone marrow. The key events for the completion of this differentiation process are the successful rearrangement and expression of the B-cell antigen receptor (BCR), consisting of Ig molecules specialized for the expression on the cell surface. The BCR on immature B cells belongs to the IgM subclass. The B cell completes its maturation process by expressing a second class of Ig, designated IgD. The IgM–IgD double-positive mature B cell leaves the bone marrow and circulates through blood and lymph, homing to secondary lymphoid organs, like the spleen and lymph nodes. B lymphocyte responses are initiated by the binding of antigen to the BCR, an event that triggers signalling cascades resulting in the transcription of a variety of genes associated with B-cell activation. In a complex interaction with other white blood cells, this activation process leads to the formation of a germinal centre (GC) reaction within secondary lymphoid organs that culminates in the transformation of the mature B cell into a terminally differentiated plasma cell (PC), whose only immunological function is the production of neutralizing soluble Igs [1]. In humans, protective antibody titres can last for >20 years, and in mice, high titres of specific antibodies are maintained for >1 year, which means protection for nearly a murine lifetime [2]. Since antibodies have a limited half-life [3, 4] continuous production of antibodies is required to warrant a protective titre. Recently, it could be shown that persistent antibody titres can be provided by long-lived PCs [5]. Thus, whereas membrane-Ig (mIg)-expressing classical memory B cells guarantee an efficient secondary immune response after contact with antigen, long-lived PCs, secreting Ig independent of antigen, represent a second tier of immunological memory.
Plasma cells and their role in humoral immunological memory
It is generally accepted that the switch of B cells expressing membrane-bound Igs, which serve as antigen receptors, to antibody-secreting plasmablasts and finally non-dividing, long-lived PCs, marks the terminal differentiation of a B cell. Some populations of long-lived PCs persist in the spleen, but most of them travel to the bone marrow or invade inflamed tissues, where they survive up to several months as resident, immobile cells in survival niches [6, 7]. However, the lifespan of PCs is limited by the immigration of newly formed migratory plasmablasts that compete with old PCs for the survival niches. In their survival niches, resident long-lived PCs are resistant to therapies targeting the activated and/or the proliferative state of lymphocytes, for example, radiation [8] and cyclophosphamide [9] treatment. The permanent and antigen-independent secretion of antibodies specific for allergens or autoantigens makes these cells key players in allergic and autoimmune diseases and obviously key targets for possible therapeutic interference.
Upon PC differentiation, there is a marked increase in the amounts of Ig mRNA transcripts, accompanied by an increase of the secretory to mIg ratio of these transcripts, as determined by differential use of poly(A) sites [10]. Several B-cell-specific markers are down-regulated upon plasma cell differentiation, including major histocompatibility complex class II, CD19, CD21, CD22 and CD45 [11]. In contrast, the proteoglycan syndecan-1 (syn-1, also designated CD138), recognizing extracellular matrix and growth factors, is up-regulated and serves as an identifying PC surface marker protein. In addition, the chemokine receptors CXCR5 and CCR7 are decreased on PCs, which impairs their migration to B- and T cell zones. On the other hand, the chemokine receptor CXCR4, which guides the PCs into CXCL12-expressing organs, including splenic red pulp, lymph nodes and the bone marrow, is highly expressed [12]. Further, it was shown that in autoimmune mice PCs not only migrate to the bone marrow but also to sites of inflammation [13]. It has long been a matter of debate as to whether the life span of PCs is intrinsically determined or dependent on several environmental factors. Recent publications now show that the longevity of the PC is influenced by a broad panel of stimuli, including cytokines like IL-5, IL-6, TNF-α, GM-CSF and the chemokine CXCL12 (stromal cell-derived factor 1-α) [13, 14]. In addition, the contact with bone marrow stromal cells provides further adhesion-dependent signals that support PC longevity [15]. These signals are most probably mediated through VLA-4 and CD44.
The migration of plasmablasts to the bone marrow is a critical differentiation step to long-lived PCs. Chemokines and their receptors are crucially involved in the control of lymphocyte trafficking. Wilson and Butcher [16] showed that the mucosal epithelial chemokine CCL28 is up-regulated in the mammary gland during lactation and that IgA-antibody-secreting cells (ASCs) from this tissue express CCR10 and migrate to CCL28. Hoyer et al. [9] and Hauser et al. [17] showed that migratory plasmablasts lose responsiveness to many chemokines, with the exception of chemokine ligand (CXCL) 12 and CXCL9. The corresponding receptors interacting with CXCL12 and CXCL9 were identified as chemokine receptor (CXCR) 4 and CXCR3. It has recently been suggested that the chemokine receptor CXCR4 is required for normal accumulation of PCs into the bone marrow [18], as demonstrated by the increased sensitivity of CXCR4 for its ligand CXCL12. Most interestingly, Muehlinghaus et al. [19] noticed the importance of the antigen receptor in modulating the migration behaviour of classical memory B cells. They showed that CXCR3 is preferentially expressed on a fraction of human memory B cells, which also expressed mIgG1. These findings led to speculations of an isotype-specific influence of the antigen receptor on the migration behaviour of plasmablasts.
Serum immunoglobulin E expression is regulated on different levels
Compared with all other Ig classes, which are present in concentrations of micrograms to milligrams per milliliter serum, the titre of IgE is very low (nano- to micrograms per milliliter range) in the plasma of normal healthy individuals and of normal laboratory mouse strains. IgE is most prominent in epithelia and mucosae, where it is bound to specific receptors on highly potent effector cells like eosinophilic granulocytes and mastcells. In these days IgE is best known for its strong, unwanted effector functions, in the form of allergic reactions [20]. These can range from annoying, local symptoms, like hayfever, to life-threatening, systemic reactions like anaphylactic shock. This underlines the potential hazard of high systemic IgE titres.
Our knowledge of the regulation of the expression of IgE and its biological function is at best limited. We do, however, know that the production of IgE is tightly regulated, which is reflected by the fact that the steady-state serum levels of IgE in mice are three to four orders of magnitude lower when compared with IgG1. Comparing the serum concentrations of IgE with IgG4 (the homologous antibody isotype to IgG1 in the murine system) in humans, a 100-fold excess of IgG4 is present. Thus, what are the rate-limiting steps responsible for this discrepancy (Fig. 1)? First, it is the complex scheme of events leading to the Ig-isotype switch. In general, class switching to the IgE locus in the mouse is less frequent than to i.e. IgG1 [21]. Our recent observations and resulting publications [22, 23] prefer the explanation of a direct IgM to IgE class switch programme, although consecutive switching events via IgG could be observed in vitro. In humans, the occurrence of consecutive class switching was reported by Cameron et al. [24]. The authors demonstrated that SεSμ as well as SεSγ DNA switch circles are produced within nasal tissue from allergic individuals following an ex vivo allergen challenge. They additionally reported an up-regulation of Cγ4 mRNA, illustrating that sequential switching to IgE also occurred. ε germline transcription was inhibited when tissue was cultured with a combination of allergen and neutralizing Abs against IL-4 and IL-13, indicating that de novo cytokine production mediated the isotype switch. Similar observations during local IgE synthesis were reported by Takhar et al. [25, 26].
Fig. 1.
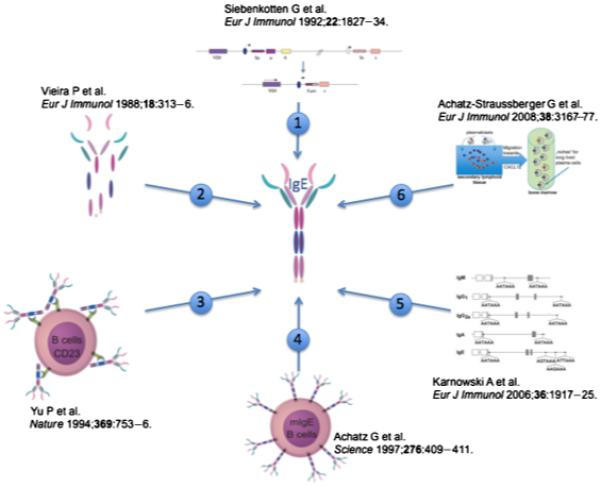
Schematic overview of B cell-specific control mechanisms, tightly regulating IgE expression in vivo. (1) Class switch recombination: although class switching to IgE and IgG1 is induced by the same cytokine milieu followed by similar signal transduction pathways, a sixfold reduced switching frequency to IgE is observed. (2) Serum half-life: IgE was reported to be degraded in 5–12 h, thus displaying the shortest half-life of all Ig isotypes. (3) Negative feed back regulation by CD23: CD23-deficient mice show a sixfold increase in serum IgE level. (4) The IgE antigen receptor: Regulation of quantity and quality of the IgE response directly correlates with the surface expression of mIgE. (5) Alternative polyadenylation: in contrast to all other isotypes, the membrane IgE (mIgE)-RNA gets polyadenylated by three cryptic, inefficient poly(A) sites. (6) Plasmablast migration: IgE plasmablasts have an intrinsic lower chance to contribute to humoral memory than IgG1 plasmablasts.
The role of IL-21 in the biology of memory B cells and plasmacells is new and intriguing, and some differences were reported between the role of IL-21 in the human and mouse immune response (for an extensive review, see [27]). In the absence of IL-21 or its receptor, mice produce little IgG1, but high levels of IgE [28]. This is not due to a defect in class-switch recombination to IgG1, and also IgG1-memory cells are reported to be present. However, in the absence of IL-21-signalling, IgG1-producing plasmacells fail to be induced [27, 28]. IL-21 can inhibit class-switch recombination to epsilon by the induction of the transcriptional inhibitor ID2 [29]. The high levels of serum IgE in IL-21- or IL-21R-deficient mice suggest further that IgE-plasmacell formation is not impaired, stressing the intrinsic differences in the various isotype-specific plasmacells. In humans, high levels of IgE are found in the hyper-IgE syndrome, which is caused by mutations in the DNA-binding domain of the transcription factor Stat3 [30, 31]. Stat3 is the most prominent transcription factor activated by IL-21 signalling, but is also involved in other signalling pathways, like IL-6- and IL-10-mediated signalling, and is involved in the activation of many cell types, e.g NK(T) cells, T cells and others, which by themselves can influence class-switch recombination, e.g. via the production of ILs and cytokines [32]. The final outcome of the process is very much dependent on local conditions, which could easily explain some of the discrepancies reported in the literature, e.g. the frequency of consecutive switching. Genetic factors also play a role: IL-21 can induce apoptosis of – activated – B cells depending on the strain: BALB/c mice are far less sensitive to IL-21-induced apoptosis than C57BL6 mice [33]. IL-21-induced apoptosis can, however, be inhibited by IL-4, illustrating the intricate interplay between cytokines and cells in the outcome of the immune response.
A further indirect regulatory mechanism is the very short half-life of serum IgE. IgE is known to exhibit the highest fractional catabolic rate. Waldmann et al. [34] put forward the hypothesis that an increased catabolic rate of IgE is dependent on the existence of intravascular and/or extravascular compartments of specifically clearing the IgE. Human IgE is reported to be metabolized mainly in the extravascular compartment, and the catabolism of IgE is related to the interaction of IgE with Fcε receptor-bearing cells. In contrast, it has also been speculated that the vascular endothelium represents a site of catabolism of IgE [34]. Serum half-lives of different Ig isotypes comparing mice and humans are summarized in Table 1.
Table 1.
Serum half-lives of different immunoglobulin isotypes
An important role in this regulatory cascade is also represented by CD23, the low-affinity receptor for IgE. In CD23 knockout mice [35], the serum levels of IgE are sixfold increased compared with wild-type mice, which implies a negative feed back regulatory mechanism on IgE expression.
A step forward in the understanding of the role of the IgE antigen receptor in the magnitude of the IgE response itself was achieved with our two mouse lines showing mutations in the membrane exons of the epsilon-heavy chain gene [36]. We showed that membrane IgE (mIgE) is the prerequisite for the production of serum IgE and that the cytoplasmic tail of mIgE plays an active role in the control of the quantity and quality of the IgE antibodies produced. With the exception of the first three amino acids, the cytoplasmic tail of mIgE shares no significant homology to cytoplasmic tails of other Ig isotypes. So far, cytoplasmic tails of Igs were not thought to have an active function in the B-cell receptor signalling. The general opinion was: Ig-tails are too short and, if at all, are able to inhibit the binding of adaptor proteins, interacting with the Igα/Igβ sheath proteins of the functional BCR. However, using the phage display technique, we were able to identify proteins, that directly interact with the cytoplasmic tail [37, 38]. This indicates the existence of an isotype (IgE)-specific signal transduction pathway, acting in coordination or even independent of the well-characterized Igα/Igβ committed pathway. So far, it is not clear whether these proteins positively or negatively influence IgE secretion. In any case, the cytoplasmic tail, or specific binding proteins or membrane-bound IgE itself, could be a target for therapeutic interference in the future.
Interestingly, mIgE expression itself is regulated by an alternative polyadenylation mechanism. While the poly(A)-signal of mRNA for serum IgE matches the consensus sequence AATAAA, three cryptic poly(A) sites with a low polyadenylation efficiency (AGTAAA, AAGAAA and ATTAAA) could be found in the 3′-UTR region of mouse mIgE [10]. This is different from the 3′-UTR regions of the other isotypes, which follow the standard consensus sequence. Comparison of the polyadenylation signal hexamers of human heavy-chain genes showed that both polyadenylation signals of the human μ and γ genes match the consensus sequence AATAAA. Those in the human ε gene are very similar to the signals in the murine ε gene: the internal polyadenylation signal matches the consensus sequence, and the three potential external polyadenylation signals (AAGAAA, AAGTAA and AAGAAA) in the 3 UTR of the human ε gene are, like in the mouse, deviant from the consensus sequence.
Based on these findings, data of our laboratory unambiguously show that the fundamental difference between IgE and other Igs lies in a poor expression of the mRNA for the membrane form of IgE. This distinct mechanism appears to set a high transcriptional threshold for mIgE expression. Because expression of the membrane form of IgE is necessary for the recruitment and survival of IgE-secreting cells, low expression of the mRNA for mIgE most likely limits the number of IgE-secreting cells and thereby limits the magnitude of an IgE-mediated immune response. Once recruited, the synthesis of IgE by PCs is comparable to that of other isotypes.
The impacts of class switching, CD23 and the IgE antigen receptor itself on the serum IgE level were reviewed in detail before [20, 39-41]. In the present review, we wish to focus on the influence of the long-lived plasma cell pool on the IgE serum level in detail.
Immunoglobulin E plasmablasts less efficiently immigrate the bone marrow
Recently, we constructed the ‘knock-in’ mouse strain KN1 [22], where we completely exchanged the membrane ε-genomic region downstream of the secretory poly(A) site for the membrane γ1-region. IgE serum titres of KN1 mice continually increased during life. In 8-week-old mice, the difference in the IgE titres between wild type (WT) and KN1 mice was not significant, but 6 months later, a four to sixfold increase of serum IgE also manifested phenotypic changes in KN1 mice. ELISpot analyses showed a correlated increase in the number of antibody-secreting cells (ASC). Therefore, the enhanced IgE level in KN1 mice was an indication of higher numbers of IgE-secreting cells. While IgE-ASCs detected in the spleen mainly belong to plasmablasts, the IgE-secreting population observed in the bone marrow most likely represented non-dividing, resident, long-lived PCs (reviewed in [42]). Our interpretation of this observation is that during plasma cell development, γ1-like signalling enhances the formation of Ig-secreting plasmablasts and long-lived PCs, reflected in KN1 mice by an overall enhanced IgE response. This implied that under normal conditions, IgG1 responses mature faster and are therefore much more efficient than IgE responses. However, once differentiated to PCs, there does not seem to be a difference between IgG1- and IgE-secreting cells (Fig. 2).
Fig. 2.
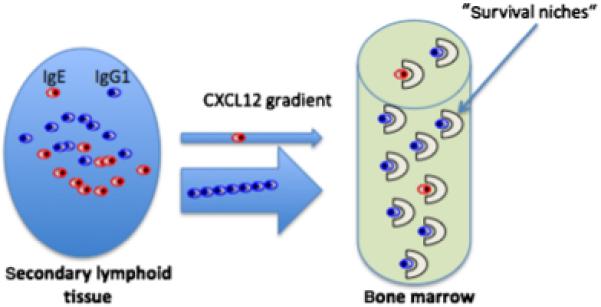
In this review, a further IgE-limiting molecular mechanism is presented. IgE-antibody-secreting cells migrate less efficiently towards the chemokine CXCL12 gradient guiding plasmablasts to plasma cell (PC) niches, than IgG1-antibody-secreting cells. Thus, IgE plasmablasts have an intrinsic, lower chance to contribute to the long-lived PC pool and thus to humoral immunologic memory than IgG1 plasmablasts.
A substantive point was observed analysing in vitro activated cultures of B cells. More CD138low, mIgE+ cells, which we consider to be similar to precursors to memory cells and early plasmablasts, arose in the KN1-derived cultures when compared with those derived from WT mice. This can be explained by a more efficient recruitment of activated B cells in this particular cell population, or by an extended lifespan of these cells. Furthermore, CD138low, mIgE+ cells of KN1 mice expressed a higher number of IgE-BCRs per cell. Of course, the increased density of BCRs bore the potential of an altered, stronger signal raised by receptor ligation. Indeed, the signal generated by the chimeric, KN1-derived IgE-BCR resembled an IgG1-driven response in that it showed an increased, late mobilization of Ca2+ ions from the extracellular space [22]. At first glance, this result might contradict the receptor ligation studies of Sato et al. [43]. The group demonstrated that ligation of IgE-BCR and IgG-BCR but not IgA-BCR generated comparable augmented Ca2+ mobilization in the B-cell line WEHI-231 as a result of a CD22-mediated signal inhibition. However, Sato and colleagues described γ2a- and ε-expressing transfectants in a cell line that is considered as representative for an immature B cell. Further, because IgG2a, in contrast to IgG1 expression, depends on Th1 cytokines, a direct comparison with our results is not warranted.
Functionally, the increased expression of the chimeric IgE-BCR on CD138low, mIgE+ cells of KN1 mice was accompanied by an increased expression of the chemokine receptor CXCR4 [22]. In view of this, it is not surprising that IgE-ASCs of KN1 mice show an increased mobility towards the ligand of CXCR4, the chemokine CXCL12. It was reported before that CXCR4 expression alone does not necessarily predict a more efficient migration behaviour [44] of ASCs. Currently, we therefore cannot exclude that factors activated in an isotype-specific fashion and different from CXCR4 further explain our observations. Because similar results were obtained when comparing the mobility towards a CXCL12 gradient of IgG1-ASCs and IgE-ASCs, we conclude that γ1-like signalling prepares early plasmablasts better for reactivity towards the chemokine CXCL12 than does ε-like signalling, conferring plasmablasts with a γ1-like signalling history a competitive edge over those with an ε-like signalling history in their quest for niches in the bone marrow.
The notion that plasmablasts matured faster to PCs under the influence of γ1-like signalling, but not ε-like signalling was additionally supported by experiments with the cytostatic drug cyclophosphamide (CycloP). In WT mice, total IgE-ASCs were more sensitive to CycloP treatment, indicating that very few IgE-ASCs had reached a fully mature, non-dividing plasma cell stage. In contrast, in KN1 mice, IgE-ASCs were more resistant to CycloP, which classifies this cell population as terminally differentiated IgE-secreting, non-dividing PCs. During an immunization experiment, we could show very clearly that a defined CycloP-resistant population of OVA-specific IgE-ASCs of KN1 mice could already be found earlier and more prominently in the bone marrow, while WT cells were rare at that time-point. We conclude that the enhanced migration behaviour of ‘γ1-signalled’ plasmablasts reflects a more efficient way to progress from the plasmablast towards the plasma cell stage.
Impact of immunoglobulin E plasmablasts on the immunoglobulin E serum level
Recently, it was suggested that IgE PCs are generated in a two-step fashion: First, in a pre-IgE phase (i.e. mIgG1-positive phase) where somatic hypermutation and affinity maturation takes place, followed by a second, IL-4-dependent phase, characterized by a switch to IgE and a swift differentiation to IgE-producing PCs [45]. At first glance, these data are compatible with our results: Erazo et al. [45] observed a fast transition of – future – IgE PCs from the GC environment to extra-GC compartments, which they claim is due to ‘the mIgG1-positive history’ of the IgE PC. Indeed we also observed a similar migration behaviour of plasmablasts that express the chimeric ε/γ1 receptor, accompanied by a rapid transition of these cells to a CycloP-resistant differentiation stage. However, we see a very different fate for the ‘true’, wild-type receptor expressing future IgE PCs: they are CycloP sensitive and show a more sluggish migratory behaviour. Further, in Erazo et al.’s [45] model, affinity maturation is solely dependent on the ‘mIgG1-positive history’ of the IgE plasma cell. From our experiments with mice with a truncated ε-cytoplasmic tail [36] and from a careful analysis of somatic mutation and affinity maturation in the IgE compartment [23], we drew very different conclusions: IgE PCs do have a mIgE-positive history and under normal conditions the recruitment of IgE PCs from mIgG1-positive memory cells is negligible. Finally, Erazo et al.’s model is a very artificial model, in which immune responses are driven by monoclonal, antigen-specific T and B cells. It must be assumed that a very artificial ‘mix’ of cytokines and ILs is produced, which most likely skews the results significantly.
Summarizing, our data strongly support the concept that BCR-mediated signalling continues in the plasmablast stage and has an isotype-specific component: γ1-like signalling, in contrast to ε-like signalling, facilitates migration of plasmablasts towards the chemokine CXCL12, allowing them to settle in plasma cell niches, like the bone marrow, and (thus) more efficiently induces progression towards the fully matured plasma cell stage. Our observations also lead to the conclusion that during a T-helper type 2 (Th2)-mediated immune response, in normal, WT mice, IgE plasmablasts have an intrinsic, lower chance to contribute to the long-lived plasma cell pool and thus to humoral immunologic memory than IgG1 plasmablasts.
Conclusion
Secreted antibodies provide specific humoral immunity. The biology of PCs (reviewed by [42]), which secrete these antibodies, remains enigmatic and is a matter of intense focus of research. What seems to be clear is the fact that a special class of PCs, the long-lived PC, immigrates to the bone marrow, forming a pool of memory PCs that is different from the pool of classical memory B cells (reviewed by [46]).
Analysing mIgE B cells is no bed of roses. The very restricted number of mIgE-expressing cells (on the average 0.04% of mouse splenic B cells) even justifies the question: are mIgE B cells necessary at all? On the one hand, there are human data to suggest that classical memory IgE B cells are almost absent because they are bypassed by a consecutive class-switch through mIgG4 [47]. On the other hand, others and we published mouse knock-out data, describing the isotype-specific immune response against the background of a missing IgE- or IgG1-antigen receptor [36, 48]. Both groups clearly demonstrated that the mIg-expressing B cell is the prerequisite for the production of serum Igs. IgE [36] and IgG1 [48] serum Ig levels were decreased by >90% in these mice. Interestingly, mIgG1 knock-out mice showed a normal IgE response and this in vivo observation thus supports the interpretation that a consecutive switch via IgG1 is not the major route of class switching in mice.
IgE-secreting long-lived PCs represent the second tier of IgE-memory and can also be detected in the bone marrow in minimal numbers (on average, 0.001% of mouse bone marrow cells) [22]. IgE PCs migrate to the bone marrow in response to CXCL12. We could show that this response is impaired for IgE-antibody-secreting cells and conclude that during a Th2-mediated immune response, in normal, WT mice, IgE plasmablasts have an intrinsic, lower chance to contribute to the long-lived PC pool and thus to humoral immunologic memory than IgG1 plasmablasts.
Apparently, an IgE immune response is, in all stages of the response, less efficiently regulated, leading to a poor response. The IgE antibodies may have strong effector functions, but the IgE response is slow and limited in developing memory responses.
Acknowledgements
Experimental work and publication charges were supported by the Austrian Science Foundation Hertha Firnberg Fellowship T166, and project P19017-B13, the Austrian National Bank (OENB grant: 11710) and the Christian Doppler Laboratory for Allergy Diagnosis and Therapy.
References
- 1.McHeyzer-Williams MG. B cells as effectors. Curr Opin Immunol. 2003;15:354–61. doi: 10.1016/s0952-7915(03)00046-3. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 3.Haba S, Ovary Z, Nisonoff A. Clearance of IgE from serum of normal and hybridoma-bearing mice. J Immunol. 1985;134:3291–7. [PubMed] [Google Scholar]
- 4.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. 1988;18:313–6. doi: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 5.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–72. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 6.Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int Immunol. 1998;10:1703–11. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- 7.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–4. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 8.Holt PG, Sedgwick JD, O’Leary C, Krska K, Leivers S. Long-lived IgE- and IgG-secreting cells in rodents manifesting persistent antibody responses. Cell Immunol. 1984;89:281–9. doi: 10.1016/0008-8749(84)90330-7. [DOI] [PubMed] [Google Scholar]
- 9.Hoyer BF, Moser K, Hauser AE, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;199:1577–84. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karnowski A, Achatz-Straussberger G, Klockenbusch C, Achatz G, Lamers MC. Inefficient processing of mRNA for the membrane form of IgE is a genetic mechanism to limit recruitment of IgE-secreting cells. Eur J Immunol. 2006;36:1917–25. doi: 10.1002/eji.200535495. [DOI] [PubMed] [Google Scholar]
- 11.Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. 2001;2:1103–8. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- 12.Cyster JG. Homing of antibody secreting cells. Immunol Rev. 2003;194:48–60. doi: 10.1034/j.1600-065x.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 13.Cassese G, Lindenau S, de Boer B, et al. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;31:2726–32. doi: 10.1002/1521-4141(200109)31:9<2726::aid-immu2726>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 14.Cassese G, Arce S, Hauser AE, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003;171:1684–90. doi: 10.4049/jimmunol.171.4.1684. [DOI] [PubMed] [Google Scholar]
- 15.Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169:4213–21. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- 16.Wilson E, Butcher EC. CCL28 controls immunoglobulin (Ig)A plasma cell accumulation in the lactating mammary gland and IgA antibody transfer to the neonate. J Exp Med. 2004;200:805–9. doi: 10.1084/jem.20041069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauser AE, Muehlinghaus G, Manz RA, et al. Long-lived plasma cells in immunity and inflammation. Ann NY Acad Sci. 2003;987:266–9. doi: 10.1111/j.1749-6632.2003.tb06059.x. [DOI] [PubMed] [Google Scholar]
- 18.Hargreaves DC, Hyman PL, Lu TT, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muehlinghaus G, Cigliano L, Huehn S, et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105:3965–71. doi: 10.1182/blood-2004-08-2992. [DOI] [PubMed] [Google Scholar]
- 20.Gould HJ, Sutton BJ, Beavil AJ, et al. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 21.Siebenkotten G, Esser C, Wabl M, Radbruch A. The murine IgG1/IgE class switch program. Eur J Immunol. 1992;22:1827–34. doi: 10.1002/eji.1830220723. [DOI] [PubMed] [Google Scholar]
- 22.Achatz-Straussberger G, Zaborsky N, Konigsberger S, et al. Migration of antibody secreting cells towards CXCL12 depends on the isotype that forms the BCR. Eur J Immunol. 2008;38:3167–77. doi: 10.1002/eji.200838456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luger E, Lamers M, Achatz-Straussberger G, et al. Somatic diversity of the immunoglobulin repertoire is controlled in an isotype-specific manner. Eur J Immunol. 2001;31:2319–30. doi: 10.1002/1521-4141(200108)31:8<2319::aid-immu2319>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 24.Cameron L, Gounni AS, Frenkiel S, Lavigne F, Vercelli D, Hamid Q. S epsilon S mu and S epsilon S gamma switch circles in human nasal mucosa following ex vivo allergen challenge: evidence for direct as well as sequential class switch recombination. J Immunol. 2003;171:3816–22. doi: 10.4049/jimmunol.171.7.3816. [DOI] [PubMed] [Google Scholar]
- 25.Takhar P, Corrigan CJ, Smurthwaite L, et al. Class switch recombination to IgE in the bronchial mucosa of atopic and nonatopic patients with asthma. J Allergy Clin Immunol. 2007;119:213–8. doi: 10.1016/j.jaci.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 26.Takhar P, Smurthwaite L, Coker HA, et al. Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J Immunol. 2005;174:5024–32. doi: 10.4049/jimmunol.174.8.5024. [DOI] [PubMed] [Google Scholar]
- 27.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 28.Ozaki K, Spolski R, Feng CG, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–4. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 29.Kishida T, Hiromura Y, Shin-Ya M, et al. IL-21 induces inhibitor of differentiation 2 and leads to complete abrogation of anaphylaxis in mice. J Immunol. 2007;179:8554–61. doi: 10.4049/jimmunol.179.12.8554. [DOI] [PubMed] [Google Scholar]
- 30.Levy DE, Loomis CA. STAT3 signaling and the hyper-IgE syndrome. N Engl J Med. 2007;357:1655–8. doi: 10.1056/NEJMe078197. [DOI] [PubMed] [Google Scholar]
- 31.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 32.Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol. 2008;19:311–8. doi: 10.1016/j.semcdb.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin H, Carrio R, Yu A, Malek TR. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J Immunol. 2004;173:657–65. doi: 10.4049/jimmunol.173.1.657. [DOI] [PubMed] [Google Scholar]
- 34.Waldmann TA, Iio A, Ogawa M, McIntyre OR, Strober W. The metabolism of IgE. Studies in normal individuals and in a patient with IgE myeloma. J Immunol. 1976;117:1139–44. [PubMed] [Google Scholar]
- 35.Yu P, Kosco-Vilbois M, Richards M, Kohler G, Lamers MC. Negative feedback regulation of IgE synthesis by murine CD23. Nature. 1994;369:753–6. doi: 10.1038/369753a0. [DOI] [PubMed] [Google Scholar]
- 36.Achatz G, Nitschke L, Lamers MC. Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science. 1997;276:409–11. doi: 10.1126/science.276.5311.409. [DOI] [PubMed] [Google Scholar]
- 37.Geisberger R, Prlic M, Achatz-Straussberger G, et al. Phage display based cloning of proteins interacting with the cytoplasmic tail of membrane immunoglobulins. Dev Immunol. 2002;9:127–34. doi: 10.1080/1044667031000137584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberndorfer I, Schmid D, Geisberger R, et al. HS1-associated protein X-1 interacts with membrane-bound IgE: impact on receptor-mediated internalization. J Immunol. 2006;177:1139–45. doi: 10.4049/jimmunol.177.2.1139. [DOI] [PubMed] [Google Scholar]
- 39.Achatz G, Achatz-Straussberger G, Luger E, Lamers R, Crameri R. Regulation of the IgE response at the molecular level: impact on the development of systemic anti IgE therapeutic strategies. Chem Immunol Allergy. 2006;91:204–17. doi: 10.1159/000090283. [DOI] [PubMed] [Google Scholar]
- 40.Achatz G, Lamers MC. The role of mIgE during thymus-dependent and thymus-independent immune responses. Allergy. 1999;54:1015–21. [PubMed] [Google Scholar]
- 41.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–17. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 42.Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–50. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 43.Sato M, Adachi T, Tsubata T. Augmentation of signaling through BCR containing IgE but not that containing IgA due to lack of CD22-mediated signal regulation. J Immunol. 2007;178:2901–7. doi: 10.4049/jimmunol.178.5.2901. [DOI] [PubMed] [Google Scholar]
- 44.Kabashima K, Haynes NM, Xu Y, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med. 2006;203:2683–90. doi: 10.1084/jem.20061289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erazo A, Kutchukhidze N, Leung M, et al. Unique maturation program of the IgE response in vivo. Immunity. 2007;26:191–203. doi: 10.1016/j.immuni.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zinkernagel RM, Bachmann MF, Kundig TM, Oehen S, Pirchet H, Hengartner H. On immunological memory. Annu Rev Immunol. 1996;14:333–67. doi: 10.1146/annurev.immunol.14.1.333. [DOI] [PubMed] [Google Scholar]
- 47.Aalberse RC, Platts-Mills TA. How do we avoid developing allergy: modifications of the TH2 response from a B-cell perspective. J Allergy Clin Immunol. 2004;113:983–6. doi: 10.1016/j.jaci.2004.02.046. [DOI] [PubMed] [Google Scholar]
- 48.Kaisho T, Schwenk F, Rajewsky K. The roles of gamma 1 heavy chain membrane expression and cytoplasmic tail in IgG1 responses. Science. 1997;276:412–5. doi: 10.1126/science.276.5311.412. [DOI] [PubMed] [Google Scholar]