

Abstract
The use of molecular modeling in conjunction with site-directed mutagenesis has extensively been used to study substrate orientation within cytochrome P450 active sites, and to identify potential residues involved in positioning and catalytic mechanisms of these substrates. However, because docking studies utilize static models to simulate dynamic P450 enzymes, the effectiveness of these studies are highly dependent on accurate enzyme models. This study employed a cytochrome P450 3A4 (CYP3A4) crystal structure (PDB code:1W0E) to predict the sites of metabolism of the known CYP3A4 substrate raloxifene. In addition, partial charges were incorporated into the P450 heme moiety to investigate the effect of the modified CYP3A4 model on metabolite prediction with the ligand-docking program Autodock. Dehydrogenation of raloxifene to an electrophilic di-quinone methide intermediate has been linked to the potent inactivation of CYP3A4. Active site residues involved in the positioning and/or catalysis of raloxifene supporting dehydrogenation were identified with the two models, and site-directed mutagenesis studies were conducted to validate the models. The addition of partial charges to the heme moiety increased accuracy of the docking studies, increasing the number of conformations predicting dehydrogenation, and facilitating the identification of substrate/active site residue interactions. Based on the improved model, the Phe215 residue was hypothesized to play an important role in orienting raloxifene for dehydrogenation through a combination of electrostatic and steric interactions. Substitution of this residue with glycine or glutamine significantly decreased dehydrogenation rates without concurrent changes in the rates of raloxifene oxygenation. Thus, the improved structural model predicted novel enzyme/substrate interactions that control the selective dehydrogenation of raloxifene to its protein-binding intermediate.
Cytochrome P450 3A4 (CYP3A4) is the most abundant human P450 found predominately in the liver and intestine, and is responsible for the metabolism of several endogenous compounds and a wide variety of xenobiotics compounds, including greater than 50% of drugs on the market (1). CYP3A4 catalyzes this metabolism by a variety of biochemical reactions, including hydroxylation, epoxidation, dealkylation, and dehydrogenation (desaturation) (2, 3), with the potential to employ multiple reactions on the same substrate. Although the majority of CYP3A4-catalyzed reactions produce inactive and nontoxic metabolites through the oxygenation of substrates, occasionally the less common dehydrogenation reaction generates highly reactive electrophiles that can form intracellular protein or DNA adducts, resulting in toxicities (4-7). Many of these electrophilic intermediates are so reactive they act as mechanism-based inactivators of the metabolizing enzyme by alkylating the prosthetic heme or binding to the apoprotein, or both (8). Although we have a general understanding about how these dehydrogenated products are formed, the precise mechanisms or factors that direct dehydrogenation versus oxygenation are not well understood. However, it is believed that steric and electronic parameters of the enzyme's active site residues facilitate dehydrogenation by positioning the substrate and/or stabilizing transition-states (9, 10). Because CYP3A4 is the major drug metabolizing P450, elucidating the factors that control CYP3A4-mediated dehydrogenation is crucial to the development of future drugs, with reduced risks of metabolic activation.
Molecular models are valuable tools to study substrate orientation within the P450 active site (11-14). However, the predictive power of molecular modeling is highly dependent on an accurate three-dimensional structure of the target (i.e. crystal structure) (15, 16). Furthermore, the force field parameters of the target are critical to accurate determinations of the interactions between a compound and its target, and to determine the lowest energy conformations and orientations of substrates in the target site (17). Unfortunately, until recently, force field parameters for the full P450 heme moiety have not been published, and therefore, have been largely ignored in docking studies involving P450s (18). This study utilized an x-ray crystal structure of CYP3A4 to model the metabolism of raloxifene, a CYP3A4-mediated dehydrogenated substrate. Furthermore, we investigated the effects of heme partial charge assignments on the predictive power of our molecular modeling.
Raloxifene presents a useful tool for studying dehydrogenation, because previous studies have shown that CYP3A4 regiospecifically dehydrogenates raloxifene to a reactive di-quinone methide, and oxygenates it to a hydroxylated metabolite (Scheme 1) (19-23). Our recent studies (23) identified an unique pathway (Scheme 1) for the production of the 7-hydroxy “metabolite” of raloxifene, through binding of the di-quinone methide to a protein carboxylic acid residue. Hydrolysis of the protein-bound ester by acidic workup liberated 7-hydroxyraloxifene, which was therefore shown to be an artifact rather than a true metabolite, formed by P450 turnover. Molecular modeling studies with raloxifene lead to the identification of several active site residues with the potential to affect substrate orientation and catalysis. However, alternative CYP3A4 crystal structures used for molecular modeling can dramatically alter substrate orientations, and predictions of enzyme/substrate interactions (15). Therefore, site-directed mutagenesis was performed to validate the models and elucidate the roles of these active site residues in substrate dehydrogenation.
Scheme 1.

Primary metabolites of CYP3A4-mediated metabolism of raloxifene
This study demonstrates the need for accurate force field parameters of the P450 active site, which is improved by the incorporation of representative partial charges on the heme moiety. The validity of the models was tested by site-directed mutagenesis studies of substrate/active site residue interactions predicted based on these docking studies. Our results demonstrate the utility of these combined techniques to evaluate the intricate interactions between dehydrogenated substrates and P450 enzymes.
Materials and Methods
Materials
Raloxifene, testosterone, 6β-hydroxytestosterone, 11β-hydroxytestosterone, NADPH, and reduced glutathione were purchase from Sigma-Aldrich (St. Louis, MO). 7-OHRA and 3′-OHRA synthesized standards (20) were generous gifts from Dr. Judy L. Bolton (University of Illinois, Chicago). All other chemicals for synthesis or analysis were of analytical grade or equivalent and obtained at the highest grade commercially available.
Instrumentation
High-performance liquid chromatography (HPLC) was conducted on an Agilent 1100 system (Agilent Technologies, Inc., Palo Alto, CA) including an autosampler and a diode-array UV/VIS detector. Chromatography was performed on a Phenomenex Luna 5μ C18 (250 × 4.60 mm) reverse-phase column (Phenomenex Inc., Torrance, CA). For testosterone metabolite analysis, the mobile phase consisted of solvent A: acetonitrile and solvent B: 1 mM ammonium acetate, with the solvent gradient program set as: 0 min, 10% A; 7 min, 35% A; 14 min, 50% A; 17 min, 55% A; 21 min, 95% A; 24 min, 95% A; and 32 min, 10% A, with a flow rate of 1 ml/min. Testosterone metabolites were monitored by UV absorption at 254 nm, and identified by comparison to standards.
LC/MS was conducted using a Thermo LCQ Advantage MAX mass spectrometer, coupled with an LC system consisting of a Finnegan Surveyor LC pump and Surveyor Autosampler (Thermo Fisher Scientific, Waltham, MA). ESI with positive ionization was utilized. The source temperature was set to 250°C, ionization voltage to 5 kV, capillary voltage 45 V, and sheath gas (N2) flow rate of 50 units. Parameters for MS/MS by CID with helium gas were as follows: activation amplitude at 35.0%, activation Q at 0.250, activation time at 30 ms and isolation width of 2 amu. Chromatography was conducted using a Phenomenex Gemini 3μ C6-Phenyl (150 × 2.00 mm) reverse-phase column (Phenomenex Inc., Torrance, CA). The mobile phase consisted of solvent A: acetonitrile and solvent B: 10% methanol and 0.4% formic acid (v/v/v). For raloxifene analysis, the mobile phase was linear from 5 to 20% solvent A over 40 min, increasing to 95% solvent A over 10 min, with a flow rate of 0.2 ml/min. Identification of raloxifene metabolites was based on [M+H]+ ion peaks as follows: hydroxyraloxifene 490 m/z; SG-raloxifene 779 m/z; SG-hydroxyraloxifene 795 m/z; and di-SG-hydroxyraloxifene 550 m/z [M+2H]2+. 7-hydroxyralxoifene and 3′-hydroxyraloxifene were also identified by comparison of retention times to standards.
Molecular docking
AutoDock 3.05 was obtained from Scripps Research Institute (La Jolla, CA). In AutoDock, the substrates raloxifene and 2-(4-hydroxy-phenyl)-benzothiophene-6-ol were treated as flexible ligands by modifying their rotatable torsions, but the CYP3A4 template was considered to be a rigid receptor. Three-dimensional coordinates of the CYP3A4 structure 1W0E (24) were acquired from the Protein Data Bank, and a second template was created (hereafter referred to as 1W0E_Modified) by assigning partial charges to the heme moiety of 1W0E. The van der Waals non-bonded parameters for these values were taken from the AMBER Parm99 force field, so that the heme force field would be compatible with AMBER protein force fields. The Fe, and all atoms bound to Fe, used the values distributed in the AMBER package. The non-bonded parameters of heme were not optimized, just the force constants and the atom charges. The structures of the substrates were built using Chem3D Ultra10 (CambridgeSoft Corporation, Cambridge, MA), and their energy was minimized using the molecular mechanics method (MM2), which was then modified by AutoDock-Tools program (Scripps Research Institute) with the Gasteiger atomic charges assigned and flexible torsions defined. The templates 1W0E and 1W0E_Modified were initially modified for docking by manually removing all water molecules. The program does not incorporate water molecules in the simulations. The polar hydrogens, protonated histidines, de-protonated aspartic and glutamic acids, Kollman partial charges, and solvation parameters used default values, assuming a pH of 7.4, with the AutoDockTools options. However, all residues were checked to guarantee that the program assigned the correct charges to each ionizable side chain. To define the active site space in which the substrates moved, AutoGrid 3.06 (Scripps Research Institute) was performed, which precalculates grids of van der Waals, hydrogen bonding, electrostatics, torsion, and solvation interactions between the templates and substrates (25). The van der Waals parameters for the heme iron atom were manually assigned Rii and epsii values of 1.3 and 0.01, respectively. A grid box with sufficient space to cover the whole active site of CYP3A4 centered at 60.795, 79.686, and 11.471; with dimensions of 52 × 62 × 68 and a resolution of 0.375 Å in each dimension. Docking was accomplished on a Dell (Round Rock, TX) Precision 690 workstation with two 64-bit Dual-Core Intel (Santa Clara, CA) Xeon processors and Red Hat (Raleigh, NC) Enterprise Linux WS4 operating system. The maximum number of energy evaluations (i.e., energy calculation of the fit of a ligand conformation) and generations (i.e., new population of individual ligand conformations) was set at 250,000 and 27,000, respectively. The rates of gene mutation and crossover rate were set at 0.02 and 0.80, respectively. The rest of the parameters were set at their default values. AutoDock searched the globally optimized conformations and orientations using the Lamarckian genetic algorithm (LGA), a hybrid of a genetic algorithm with an adaptive local search (LC) method (25). One hundred LGA runs were conducted for each template and docking solutions were groups of conformational clusters where all-atom root mean square deviations (RMSD) within 2.0 Å of each other were clustered together, using AutoDockTools. Thus, clusters were grouped together based on their geometric similarities, not their energy similarities.
CYP3A4 mutant construction
CYP3A4 mutants S119A, F215G, F215Q, and T309V were generated by polymerase-chain reaction (PCR) using QuikChange® XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). The CYP3A4 containing construct, pSE3A4His (26), was used as the template, and the forward and reverse primers are shown in Table 1. All mutant constructs were sequenced to confirm correct mutations and lack of anomalous mutations (DNA Sequencing and Genomics Core Facility, University of Utah, Salt Lake City, UT). The pSE3A4His construct was a generous gift from Dr. James R. Halpert (University of California, San Diego, CA).
Table 1.
Primers used for generation of CYP3A4 mutants. Mutated nucleotides are highlighted.
| Mutant | Primers |
|---|---|
| S119A | 5′-gatttatgaaaagtgccatcgctatagctgaggatgaagag-3′ |
| 5′-ctcttcatcctcagctatagcgatggcacttttcataaatc-3′ | |
| T309V | 5′-ttatctttatttttgctggctatgaagtcacgagcagtgttctct-3′ |
| 5′-agagaacactgctcgtgacttcatagccagcaaaaataaagataa-3′ | |
| F215G | 5′-gaacaccaagaagcttttaagatttgatggtttggatccattctttctctcaataac-3′ |
| 5′-gttattgagagaaagaatggatccaaaccatcaaatcttaaaagcttcttggtgttc-3′ | |
| F215Q | 5′-tttgtggagaacaccaagaagcttttaagatttgatcaattggatccattctttctct-3′ |
| 5′-agagaaagaatggatccaattgatcaaatcttaaaagcttcttggtgttctccacaaa-3′ | |
Enzyme expression
CYP3A4 and mutants were expressed in Escherichia coli DH5-α cells (Invitrogen, Carlsbad, CA) and purified as described previously (26). Rat cytochrome P450-oxidoreductase (POR) construct pOR262, which was a generous gift from Dr. Charles B. Kasper (University of Wisconsin, Madison, WI), was expressed and purified in JM-109 cells (Stratagene, La Jolla, CA) as described previously (27). The P450 content was determined by reduced carbon monoxide difference spectra (28).
Testosterone activity assay
The reconstituted system contained 50 pmol of purified P450, 100 pmol of recombinant POR, 100 pmol of cytochrome b5 (Invitrogen, Carlsbad, CA), 0.04% sodium cholate, and 20 μg of lipid mix (equal weights of DOPC, DLPC and DLPS). The mixture was gently shaken at room temperature for 10 min. To the system was added potassium phosphate buffer (50 mM, pH 7.4), GSH (4 mM), MgCl2 (15 mM), and testosterone (25-400 μM) in a final volume of 500 μl. The mixture was preincubated at 37°C for 5 min, and the reaction was initiated by the addition of 2 mM NAPDH. The reaction was allowed to proceed for 10 min at 37°C and then terminated by the addition of 500 μl of ice cold methanol containing 2.5 μM of the internal standard 11β-hydroxytestosterone. The mixtures were vortexed, followed by centrifugation at 21000 × g for 15 min to remove the protein. The organic components of the supernatant were concentrated to 100 μl under nitrogen gas for analysis via HPLC. The amount of 6β-hydroxytestosterone formed at each testosterone concentration was determined with a standard curve and used for kinetic analysis. Kinetic parameters including Vmax, the maximal velocity, and Km, the substrate concentration at half-maximal velocity, were obtained by a nonlinear least-squares regression fitting of the Michaelis-Menten equation, V = Vmax × [S]/(Km + [S]), using the “Solver” function from Microsoft Excel 2003 (Microsoft, Redmond, WA). All incubations were done in triplicate (i.e., 3 separate reconstituted systems).
Raloxifene dehydrogenation assay
The reconstituted system was the same as described above, with the exception that raloxifene (25 μM) was added to the incubations. The mixture was preincubated at 37°C for 5 min, and the reaction was initiated by the addition of 2 mM NAPDH. The reaction was allowed to proceed for 10 min at 37°C and then terminated by addition of 100 μl of 60% trichloroacetic acid (v/v). The mixtures were vortexed, followed by centrifugation at 21000 × g for 15 min to remove the protein. The organic components of the supernatant were extracted using C-18 Sep-Pak cartridges (Waters, Taunton, MA). The acetonitrile eluate was concentrated to dryness by evaporation under nitrogen and reconstituted in 10% acetonitrile/ H2O (v/v) for analysis via LC/MS. All incubations were repeated 5 times (i.e., 5 separate reconstituted systems).
Results
Molecular docking of substrates into 1W0E and 1W0E_Modified
Until recently, the charge state parameters for the heme moiety of P450s had not been defined, and therefore were set to zero by most docking software. As a result, the heme's electronic contribution to docking ligands into the active site of P450s was largely overlooked. To investigate the effects of heme parameters, raloxifene was docked into the templates 1W0E (with no heme partial charges parameters) and 1W0E_Modified (with partial charges added to the heme). One hundred docking conformations for each CYP3A4 crystal structure were clustered with a RMSD of 2.0 Å and ranked by the lowest docking energy for analysis (Figure 1 A&B). Raloxifene is metabolized by CYP3A4 to two primary metabolites the di-quinone methide and 3′-hydroxyraloxifene (Scheme 1). The di-quinone methide is a dehydrogenated product, most likely initiated by hydrogen abstraction from the hydroxyl group on C-6 of the benzothiophene moiety. The other major raloxifene metabolite is hydroxylated on the C-3′ atom, possibly initiated by hydrogen abstraction from the hydroxyl group on C-4′ of the phenol or π-bond oxygenation. To score our docking results for predictions of the sites of metabolism, any conformation where the hydroxyl group on the benzothiophene moiety was within 5 Å of the heme iron was considered a successful prediction for raloxifene dehydrogenation. Likewise, any conformation where the phenol's hydroxyl group was within 5 Å of the heme iron was considered a successful prediction for raloxifene 3′-hydroxylation. Any conformation that did not meet either criterion was considered an unsuccessful, or a metabolically “inactive” prediction (Table 2). Out of the 100 docking conformations from the 1W0E template (with no heme charge assignments), only 10% predicted raloxifene dehydrogenation, 5% predicted raloxifene 3′-hydroxylation, and 85% predicted metabolic “inactive” states (Figure 2 A&B). The lowest energy cluster predicted dehydrogenation (Figure 1A). However, this cluster only accounted for two out of 100 conformations. The largest cluster (10 conformations) from this docking study predicted metabolic “inactive” states. Out of the 100 docking conformations from the 1W0E_Modified template, 24% predicted dehydrogenation, 4% predicted 3′-hydroxylation, and 72% predicted metabolic “inactive” states (Figure 2 C&D). The lowest energy cluster, which was also the largest cluster, predicted dehydrogenation (Figure 1B).
Figure 1.
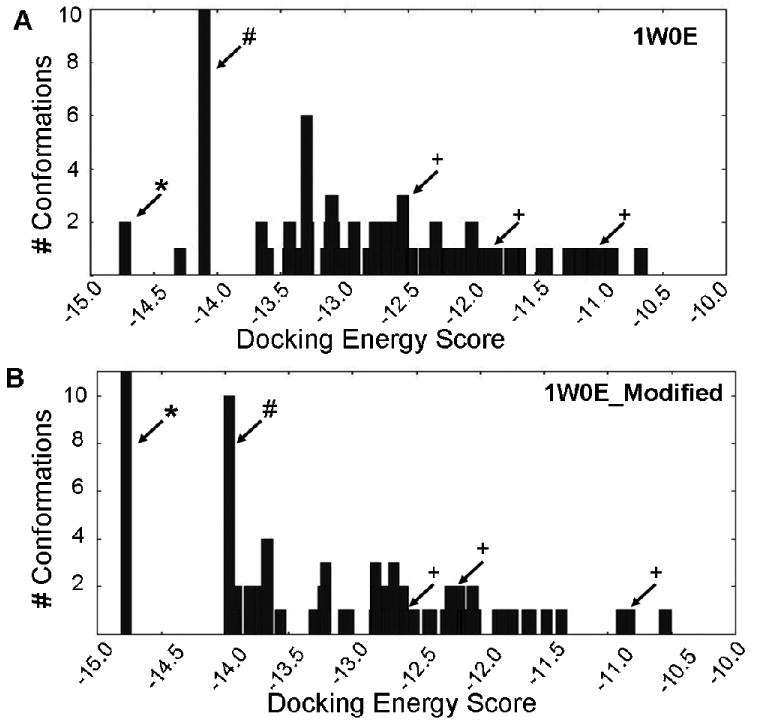
Histograms of energy clusters of 100 conformations generated by docking raloxifene into (A) 1W0E and (B) 1W0E_Modified using AutoDock 3.0. Conformations generated by AutoDock were clustered at RMSD = 2 Å, and then plotted by the lowest energy conformation of each cluster. (*) indicates most abundant/lowest energy cluster predicting raloxifene dehydrogenation. (#) indicates largest cluster that predicts “inactive” conformations. (+) indicates conformation that predicts 3′ hydroxylation of raloxifene.
Table 2.
Percentage of accurate raloxifene metabolite predictions (out of 100 conformations) from docking raloxifene into CYP3A4
| Template | Dehydrogenation | 3′ Hydroxylation | Inactive |
|---|---|---|---|
| 1W0E | 10% | 5% | 85% |
| 1W0E_Modified | 24% | 4% | 72% |
Figure 2.
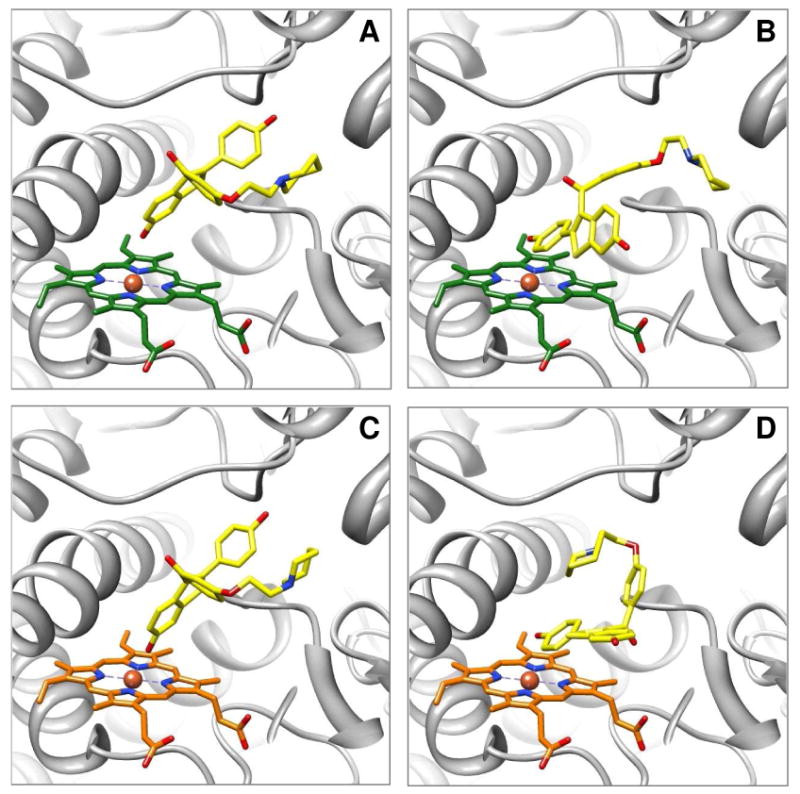
Molecular models of raloxifene in the active site of CYP3A4 predicted by AutoDock 3.0. Raloxifene was docked into 1W0E, with an unmodified heme, or into 1W0E_Modified, where partial charges were assigned to the heme. A representative conformation from the largest/lowest energy cluster, predicting dehydrogenation, from docking with 1W0E is depicted in (A), and from docking with 1W0E_Modified is depicted in (C). The dehydrogenation orientation of raloxifene is portrayed in (A) and (C), with the hydroxyl group on C-6 of the benzothiophene moiety oriented toward the heme and close to the Fe. A representative conformation predicting raloxifene 3′ hydroxylation from docking with 1W0E is depicted in (B), and from docking with 1W0E_Modified is depicted in (D). 3′ hydroxylation of raloxifene is portrayed in (B) and (D), with the hydroxyl group on C-4′ of the phenol moiety oriented toward the heme. CYP3A4 is shown in a ribbon format, iron as a sphere, heme (green for 1W0E and orange for 1W0E_Modified) and raloxifene (yellow) in color-coded sticks: nitrogen = blue, oxygen = red. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
The core structure of raloxifene, 2-(4-hydroxy-phenyl)-benzothiophene-6-ol, where CYP3A4-mediated metabolism is known to take place, was docked into 1W0E_Modified to determine if there was a preference for one side of the molecule to orient itself toward the heme. One hundred docking conformations were clustered with an RMSD of 2.0 Å and ranked by the lowest docking energy for analysis (Figure 3). Out of 100 conformations, 40% predicted metabolism on the phenol moiety, 24 % predicted metabolism on the benzothiophene moiety, and 31% predicted “inactive” states. Furthermore, the first and second most abundant/lowest energy clusters predicted metabolism at the phenol moiety, whereas the clusters that predicted metabolism at the benzothiophene moiety had higher energy (i.e., less favorable).
Figure 3.
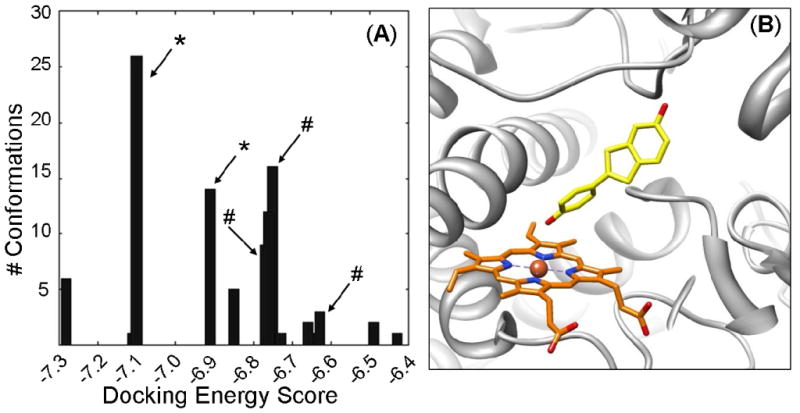
Docking of a simplified benzothiophene molecule into the CYP3A4 active site. (A) Histogram of 100 conformations from docking 2-(4-hydroxy-phenyl)-benzothiophene-6-ol into 1W0E_Modified (with heme partial charges assigned) using AutoDock 3.0. Conformations generated by AutoDock were clustered with an RMSD of 2 Å, and plotted by the lowest energy conformation of each cluster. (*) Indicates clusters with the phenol moiety oriented toward the heme. (#) Indicates cluster with the benzothiophene moiety oriented toward the heme. Unmarked clusters have “inactive” conformations. (B) A representative conformation from the most abundant/lowest energy cluster of 2-(4-hydroxy-phenyl)-benzothiophene-6-ol, which predicts that the phenol moiety is oriented toward the heme. CYP3A4 is shown in a ribbon format, iron as a sphere, heme modified with partial charges (orange) and docking 2-(4-hydroxy-phenyl)-benzothiophene-6-ol (yellow) in color-coded sticks: nitrogen = blue, oxygen = red. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Identification of Active Site Residues
Raloxifene docked into the 1W0E template only predicted raloxifene dehydrogenation for 10% of all the conformations. Furthermore, the lowest energy cluster, which predicted dehydrogenation, only accounted for two conformations. Despite the poor results obtained from docking raloxifene into 1W0E, the conformations that predicted dehydrogenation were visually analyzed to identify active site residues that were < 5 Å from raloxifene, and that could potentially position raloxifene for dehydrogenation and/or stabilize transition states. In several of the conformations, the hydroxyl group on the Thr309 side chain was within hydrogen bonding distance, approximately 3.0 Å, of the oxygen of the hydroxy-benzothiophene moiety, indicating that Thr309 could potentially stabilize a transition state or phenoxy radical intermediate, during raloxifene dehydrogenation (Figure 4A). The hydroxy group on the Ser119 side chain was within hydrogen bonding distance, approximately 3.7 Å, of the oxygen of the hydroxy-benzothiophene moiety, indicating that S119 could potentially stabilize the transition-state, or phenoxy radical intermediate, during raloxifene dehydrogenation (Figure 4B).
Figure 4.
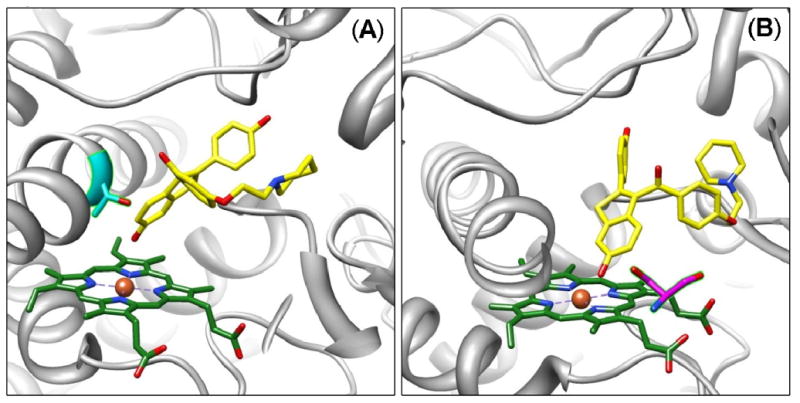
Molecular models of raloxifene in the active site of 1W0E (without heme partial charges) predicted by AutoDock 3.0. (A) A conformation predicting dehydrogenation of raloxifene, where the active site Thr309 residue (cyan) is in position to donate a hydrogen bond to the oxygen of the hydroxyl group on the benzothiophene moiety of raloxifene. (B) A conformation predicting dehydrogenation of raloxifene, where the active site Ser119 residue (magenta) is in position to donate a hydrogen bond to the oxygen of the hydroxyl group on the benzothiophene moiety of raloxifene. CYP3A4 is shown in a ribbon format, iron as a sphere, heme without partial charges (green) and docking raloxifene (yellow) in color-coded sticks: nitrogen = blue, oxygen = red. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Docking studies with 1W0E_Modified (with heme partial charges) had a dramatic effect, increasing the prediction of raloxifene dehydrogenation to 24% of all the conformations. Most remarkable was the increase in the most abundant/lowest energy cluster, which increased to 11% of all conformations. This cluster alone was visually analyzed to identify active site residues that could potentially position raloxifene for dehydrogenation and/or stabilize transition states. Phe215, located in the F-G loop (between the F and F′ helixes), appeared to play a key role in positioning raloxifene for dehydrogenation (Figure 5). The steric interactions between raloxifene and the Phe215 residue appear to provide a hydrophobic pocket where the phenol and 1-(2-phenoxy-ethyl)-piperidine moieties of raloxifene wrapped around the benzene ring of the phenylalanine side chain. In addition to the steric interactions, raloxifene's phenol moiety was positioned to enable a π bond T-stack with the phenylalanine residue. These steric and electronic interactions appeared to position the benzothiophene moiety of raloxifene towards the heme, supporting the dehydrogenation pathway.
Figure 5.
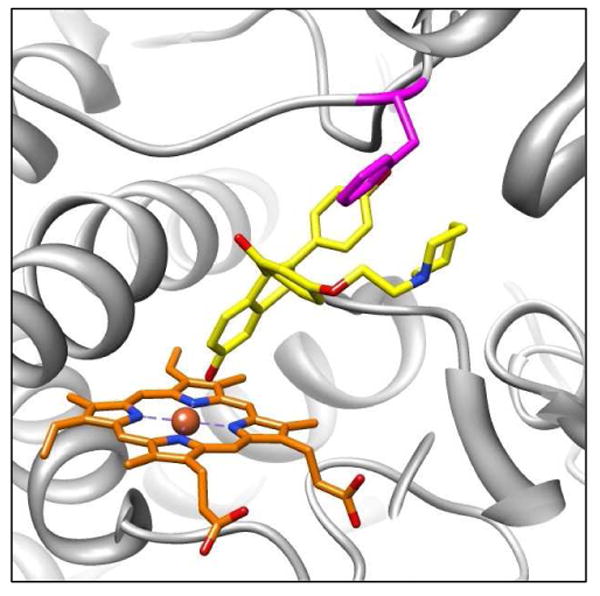
Molecular models of raloxifene in the active site of 1W0E_Modified (with partial charges assigned to the heme) predicted by AutoDock 3.0. A representative conformation from the largest/lowest energy cluster predicting the dehydrogenation of raloxifene. The active site Phe215 residue (magenta) is in position to orient raloxifene by steric interactions and π bond T-stacking between raloxifene's phenol moiety and Phe215's benzene ring. CYP3A4 is shown in a ribbon format, iron as a sphere, heme with partial charges (orange) and docking raloxifene (yellow) in color-coded sticks: nitrogen = blue, oxygen = red. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Construction and expression of CYP3A4 mutants
Molecular docking studies of raloxifene docked into CYP3A4 templates 1W0E and 1W0E_Modified were utilized to identify active site residues that potentially positioned raloxifene for dehydrogenation. From the 1W0E template, Ser 119 and Thr 309 were selected for mutation, and from the 1W0E_Modified template, Phe215 was selected for mutation. Single amino acid mutants S119A, T309V, F215G and F215Q were successfully introduced, as confirmed by full-length sequence analysis within the P450 cDNA region of the expression vector pSE3A4His. All mutants were successfully expressed, but with lower expression levels, S119A (10 nmol/L), T309V (7 nmol/L), F215G (12 nmol/L) and F215Q (10 nmol/L), than native CYP3A4 (45 nmol/L). The absence of a large 420 nm peak in the reduced carbon monoxide difference spectra suggested that the decrease in mutant protein yield was due to a decrease in expression, and not misfolding of the enzymes.
CYP3A4 mutant testosterone hydroxylation activity
To ensure that the recombinant CYP3A4 mutants were functioning enzymes, they were incubated with the CYP3A4 model substrate testosterone. Their activities were determined by monitoring 6β-hydroxytestosterone production, and Km and Vmax were calculated for each enzyme (Table 3). All of the expressed CYP3A4 enzymes were active, successfully producing 6β-hydroxytestosterone. Native CYP3A4's Km and Vmax were calculated to be 240 ± 20 μM and 21 ± 4 min-1, respectively. Both the CYP3A4 F215G and F215Q mutant enzymes demonstrated a lower affinity for testosterone with Km of 370 ± 30 μM and 320 ± 20 μM, and lower maximal enzymatic turnover of testosterone with Vmax of 16 ± 1 min-1 and 17.0 ± 1 min-1, respectively. The T309V mutation resulted in an increase of testosterone affinity, but decrease in enzymatic turnover, with a Km and Vmax of 110 ± 20 μM and 4.5 ± 0.4 min-1, respectively. The S119A mutation had a slight increase in testosterone affinity and an increase in enzymatic turnover, with a Km and Vmax of 230 ± 20 μM and 29 ± 1 min-1, respectively.
Table 3.
Kinetic constants for 6β-hydroxylation of testosterone by CYP3A4 and mutants
| Enzyme | Km (μM) | Vmax (nmol/min/nmol) | Vmax/Km |
|---|---|---|---|
| WT | 240 ± 20 | 21 ± 4 | 0.086 |
| S119A | 230 ± 20 | 29 ± 1 | 0.13 |
| T309V | 110 ± 20 | 4.5 ± 0.4 | 0.040 |
| F215G | 370 ± 30 | 16 ± 1 | 0.043 |
| F215Q | 320 ± 20 | 17 ± 01 | 0.053 |
CYP3A4 mutant raloxifene dehydrogenation activity
To investigate the effects of the single amino acid mutations on dehydrogenation, the P450 mutants were incubated with raloxifene. Due to a lack of standards for the raloxifene/GSH adducts, the metabolites were estimated with use of an internal standard via LC/MS. All the CYP3A4 mutants metabolized raloxifene, producing previously identified hydroxylated raloxifene metabolites and GSH adducts (19, 20), with no new metabolites detected. Surprisingly, despite the changes that the S119A and T309V mutations had on testosterone activity, neither mutation significantly altered raloxifene metabolism. Both CYP3A4 S119A and T309V produced the same relative amounts of raloxifene metabolites, with the same metabolic distribution, as native CYP3A4 (data not shown). The CYP3A4 F215Q mutation significantly decreased (p < 0.05) formation of 3′-hydroxyraloxifene (3′-OHRA) (30%), mono-GS-raloxifene (GS-RA) (28%), and di-GS-hydroxyraloxifene (di-GS-OHRA) (22%), compared to native CYP3A4 (Figure 6). CYP3A4 F215Q had no significant effect on the production of 7-hydroxyraloxifene (7-OHRA). The CYP3A4 F215G mutation significantly decreased (p < 0.05) production of GS-RA (40%) and di-GS-OHRA (55%), but had no significant effect on 7-OHRA or 3′-OHRA formation (Figure 6).
Figure 6.

Native CYP3A4, CYP3A4F215Q, and CYP3A4F215G production of raloxifene metabolites. The relative amounts of metabolites were estimated with an internal standard and concentration is expressed in arbitrary units (AU). Mutations of Phe215 reduced raloxifene dehydrogenation, demonstrated by the decreased production of GS-RA, the GSH adduct of the dehydrogenated product. (*) Indicates a significant difference (p < 0.05, n = 5) compared to native CYP3A4.
Discussion
Substrate orientation within the active site of P450s is a crucial factor for P450-mediated metabolism. Therefore, docking studies can be particularly useful to gain selectivity and steric information on potential compounds, which can be used to predict their sites of metabolism and possible toxic metabolites. Our previous docking studies with indapamide and two different CYP3A4 crystal structures (1TQN and 1W0E), demonstrated the importance of accurate crystal structures to improve docking fidelity (15). While both CYP3A4 crystal structures accurately predicted major pathways of indapamide metabolism, accurate predictions of substrate/active site residue interactions could not be determined because the active site residues of the two structures were different. The 1TQN template predicted that Arg212 played an important role in positioning indapamide for dehydrogenation, while 1W0E predicted that Arg212 was not oriented within the active site and did not interact with indapamide. Only after site-directed mutagenesis studies on the Arg212 residue was it determined that Arg212 was not necessary for substrate orientation to facilitate dehydrogenation of indapamide. From these results, and from examining other CYP3A4 crystal structures, it was concluded that Arg212 is most likely oriented away from the heme moiety. Therefore, the 1TQN structure did not appear to be an appropriate template for molecular docking studies, so the 1W0E crystal structure was used for this study.
In addition to the importance of a correct structure, the use of accurate force field parameters is essential for successful molecular docking studies. However, due to a lack of published heme partial charge parameters, the vast majority of past docking studies with P450s ignored the electronic contributions of this vital prosthetic group. With newly established heme force field parameters, we can incorporate their contributions to molecular docking studies involving P450s. In this study, we utilized the CYP3A4 crystal structure 1W0E, with and without the addition of heme parameters, to determine if they improved docking results. When raloxifene was docked into 1W0E (without heme partial charges), the lowest energy cluster did successfully predict raloxifene dehydrogenation. However, this cluster only represented 2% of all the conformations. Overall, only 10% of the conformations predicted raloxifene dehydrogenation. The docking study also successfully predicted raloxifene 3′-hydroxylation; however, these conformations were much higher in energy, compared to the dehydrogenation conformations (Figure 1A). The largest cluster from this docking study, representing 10% of conformations, corresponded to metabolic “inactive” conformations. Furthermore, 85% of all the conformations represented “inactive” conformations. When raloxifene was docked into 1W0E_Modified, there was significant improvement in the prediction of the site of dehydrogenation. The lowest energy cluster, which also represented the largest cluster of conformations (11%), predicted raloxifene dehydrogenation (Figure 1B). Furthermore, the total number of substrate conformations that predicted dehydrogenation increased to 24%. Interestingly, successful predictions of raloxifene 3′-hydroxylation were relatively unchanged at 4%, and again these conformations were higher in energy than the dehydrogenation conformations. The second largest cluster (10%) still predicted “inactive” conformations, and overall 75% of the conformations were “inactive.” Thus, these results demonstrated the importance of accurate heme force field parameters in molecular docking studies. While 1W0E did successfully predict raloxifene metabolites, without the benefit of prior in vitro studies, the lack of large clusters with low energy would not permit confident predictions of raloxifene metabolic pathways with this model. In contrast, the incorporation of the heme parameters into 1W0E_Modified increased the successful prediction of raloxifene dehydrogenation from 10 to 24%. Furthermore, while the largest cluster from 1W0E docking (without heme parameters) predicted “inactive” conformations, the largest and lowest energy cluster from 1W0E_Modified predicted raloxifene dehydrogenation. Docking with both models resulted in a large percentage of inactive conformations. However, this is not unexpected for CYP3A4, due to its relatively large active site and high degree of flexibility (29-31); one would expect difficulties in modeling this P450 with a static crystal structure. Even with the large number of “inactive” conformations, this approach is useful to predict raloxifene metabolites, because the vast majority of “inactive” conformations have vastly different orientations (greater that RMSD 2 Å), and therefore do not cluster together. The only large cluster of “inactive” conformations place raloxifene 6.5 Å or greater away from the heme, and in an orientation that would not produce any known metabolites.
To identify potential active site residues that may position and/or stabilize a transition state, to facilitate dehydrogenation, raloxifene was docked with the CYP3A4 crystal structure 1W0E (without heme partial charges). This study resulted in very few conformations predicting dehydrogenation at the benzothiophene moiety. However, the few conformations that predicted dehydrogenation were visually analyzed to identify potential active site residues that might interact with raloxifene. In several conformations, the hydroxy group on the benzothiophene moiety was within hydrogen bonding distance of the hydroxy group on the Thr309 residue (Figure 4A). This threonine residue is already theorized to play an important role in the formation of the reduced ferric peroxyl radical intermediate (compound 0) in the P450 catalytic cycle, by donating a hydrogen bond to stabilize the Fe-OO moiety (32). Therefore, we postulated that Thr309 could also provide a hydrogen bond, and stabilize the raloxifene dehydrogenation transition-state (i.e., donate a hydrogen bond to the phenoxyl radical of the benzothiophene moiety, after removal of a hydrogen atom from the phenol). In another conformation, the hydroxyl group on the benzothiophene moiety was within hydrogen bonding distance of the hydroxy group on the Ser119 residue (Figure 4B). Previous studies have shown that Ser119 may play a vital role in the CYP3A4-mediated metabolism of progesterone (33) and midazolam (34). Therefore, we postulated that Ser119 might also position raloxifene for dehydrogenation. To investigate the role of Thr309 and Ser119 in raloxifene metabolism, and thereby validate our docking study of raloxifene, site-directed mutagenesis was utilized to create CYP3A4 T309V and S1319A mutants. These mutations where chosen because they have similar steric features, but lack hydroxyl groups for hydrogen bonding. Based on testosterone activity, the T309V mutation decreased enzyme efficiency by 55% and the S119A mutation increased the enzyme efficiency by 50%, compared to native enzyme. Despite the effects on testosterone activity, when CYP3A4 T309V and S119A were incubated with raloxifene, these mutations had no significant effects on raloxifene metabolism. Therefore, we concluded that Thr309 and Ser119 do not affect raloxifene orientation, or stabilize transition states. From these results we also concluded that our model, based on docking raloxifene into 1W0E (without heme partial charges), was not sufficiently accurate. This was not surprising, given the poor docking results from the 1W0E template.
Previous studies on CYP3A4-mediated metabolism of raloxifene have shown that it is metabolized on both the benzothiophene and phenol moieties, located on opposite ends of the molecule (19, 20). To determine if there was preference for either end of the core structure of raloxifene to orient itself toward the heme moiety of CYP3A4, docking studies were conducted with a smaller, simplified analog, 2-(4-hydroxy-phenyl)-benzothiophene-6-ol, and the 1W0E_Modified structure. The results demonstrated that the preferred orientation of 2-(4-hydroxy-phenyl)-benzothiophene-6-ol in CYP3A4 was with the phenol moiety pointing toward the heme (Figure 3). This is surprising because previous docking studies predicted that the preferred raloxifene orientation is with the benzothiophene moiety pointed toward the heme, and incubations with CYP3A4 have shown metabolism at the benzothiophene moiety as one of the most abundant CYP3A4-mediated reactions. Therefore, it was theorized that the piperidine moiety of raloxifene must play an important role in orienting raloxifene for dehydrogenation at the benzothiophene moiety.
In contrast to the docking study with 1W0E (without heme partial charges), the 1W0E_Modified docking study produced the lowest-energy cluster with the largest population. Therefore, we utilized this cluster alone to identify potential active site residues that may position and/or stabilize a transition state, facilitating raloxifene dehydrogenation. To refine the search, and account for the results for docking the 2-(4-hydroxy-phenyl)-benzothiophene-6-ol into CYP3A4, close attention was paid to the positioning of the 4-(2-piperidine-1-ly-ethoxy)-benzaldehyde moiety. By visual analysis, we identified the Phe215 residue as an important for positioning raloxifene for dehydrogenation. In all the conformations, the phenol and piperidine moieties wrap around the Phe215 residue (Figure 5), orienting raloxifene with the benzothiophene moiety pointed toward the heme. In addition to the steric effects of Phe215, there also appeared to be π bond T-stacking between raloxifene's phenol moiety and the phenylalanine benzene ring. Phe215 belongs to a highly-ordered cluster of seven phenylalanines (Phe108, 213, 215, 220, 241 and 304) in the active site of CYP3A4, which appears to be a unique feature among all P450s that have been crystallized. Previous studies have shown that several of these phenylalanines play a role in substrate cooperativity, regioselectivity, and stereoselectivity (26, 34, 35). To investigate the role of Phe215 in raloxifene metabolism, and validate our docking study of raloxifene with 1W0E_Modified, site-directed mutagenesis was utilized to create CYP3A4 F215G and F215Q mutants. The CYP3A4 F215Q mutant was chosen to eliminate π bond T-stacking between the residue and the substrate, while keeping a bulky residue for steric interactions. The CYP3A4 F215G mutant was chosen to eliminate both the π bond T-stacking and the steric effects of the residue. The activity of these mutants was determined by metabolism of the CYP3A4 model substrate testosterone. The CYP3A4 F215G and F215Q mutations decreased the affinity for testosterone, and decreased substrate turnover, resulting in a substantial decrease in enzyme efficiency by 50% and 38%, respectively, compared to native CYP3A4. The enzymes were incubated with raloxifene to investigate the effects of the F215G and F215Q mutations on dehydrogenation. Because the dehydrogenated product of raloxifene is too unstable to measure directly (20), the mono-GSH adduct of the reactive intermediate (GS-RA) was measured to monitor raloxifene dehydrogenation. The CYP3A4 F215Q mutation significantly decreased the production of 3′-OHRA, GS-RA, and di-GS-OHRA, compared to native CYP3A4 (Figure 6). However, there was no significant effect on the production of 7-OHRA, which is theorized to be an artifact rather than a P450-catalyzed oxygenation product, formed by the conjugation of raloxifene di-quinone methide with a carboxylic acid moiety of CYP3A4 and subsequently released by acid-catalyzed hydrolysis of the ester to form 7-OHRA (23). The CYP3A4 F215G mutation produced an even more dramatic decrease in the production of GS-RA and di-GS-OHRA, but had no significant effect on 7-OHRA or 3′-OHRA (Figure 6). These results clearly demonstrated that the Phe215 residue plays a substantial role in positioning raloxifene for dehydrogenation. The F215Q mutation eliminated any possible π bond T-stacking interaction with raloxifene, but still provided a large residue to position raloxifene by steric interactions. The significant decrease in GS-RA, resulting from the F215Q mutation, suggested that π bond T-stacking between the Phe215 and the phenol moiety of raloxifene does play a role in positioning raloxifene for dehydrogenation. The F215G mutation eliminated both the π bond T-stacking interaction and the steric interaction that positions raloxifene with the benzothiophene moiety oriented toward the heme. This mutation had an even more dramatic effect on GS-RA production, demonstrating that both the steric interaction and electrostatic interaction are important to position raloxifene for dehydrogenation. Furthermore, while the F215G mutation decreased GS-RA production (i.e., dehydrogenation) by 40%, it had no significant effect on 3′-OHRA production (i.e., oxygenation). These results demonstrated that the F215G mutation specifically decreased raloxifene dehydrogenation, not just overall raloxifene metabolism. Interestingly, the CYP3A4 mutants and native enzyme produced the same amount of 7-OHRA. Because 7-OHRA and GS-RA are both formed from the same di-quinone methide intermediate, one might expect the mutant enzymes to produce less 7-OHRA. However, a possible explanation for these disparate results is that the site of carboxyl conjugation, which results in the ester that can be hydrolyzed to release 7-OHRA, is saturatable. Therefore, only a finite and equal amount of 7-OHRA can be formed from both the native and mutant enzymes. Another theory is the di-quinone methide has a higher affinity for thiol groups (i.e., GSH) than carboxyl groups. It is possible that GSH in the active site (36) could have obscured small changes is ester formation. Therefore, we could not observe an effect on production of 7-OHRA during the short incubation time period. The CYP3A4 mutants also decreased di-GS-OHRA production, compared to native CYP3A4. The di-GS-OHRA adduct may be produced from CYP3A4 oxidation of 3′-OHRA to a reactive o-quinone or a different quinone methide, which is conjugated by GSH to form GS-OHRA, and undergoes additional redox cycling or P450-mediated oxidation to form di-GS-OHRA. We opine that these results can be interpreted to mean that the Phe215 mutation decreases CYP3A4-mediated oxidation of 3′-OHRA to o-quinone or quinone methide species, rather than causing decreased formation of 3′-OHRA, followed by GSH conjugation, redox cycling, and a second GSH conjugation. Additional experiments are needed to clarify the source and fate of these secondary raloxifene metabolites.
The results of this study demonstrate the current complexities in using molecular modeling in conjunction with site-directed mutagenesis to study P450-mediated metabolism, but also exhibit the great potential for continued use and improvement of these combined techniques. Docking raloxifene into 1W0E (without heme partial charges) resulted in poor predictions for raloxifene metabolism. Not surprisingly, mutations based on docking with 1W0E (S119A and T309V) had no effect on raloxifene dehydrogenation. However, the addition of partial charges to the heme dramatically increased the accuracy of the predictions of dehydrogenation when raloxifene was docked into 1W0E_Modified. Analysis of the most abundant/lowest energy cluster from this study identified the Phe215 residue as an important component of the enzyme tertiary structure that would interact with raloxifene. Site-directed mutagenesis experiments with Phe215 clearly demonstrated that Phe215 positions raloxifene for dehydrogenation, most likely through a combination of π bond T-stacking and steric interactions. Furthermore, these results validate the 1W0E_Modified model, and demonstrate the usefulness of molecular modeling in conjunction with site-directed mutagenesis to study P450-mediated mechanisms.
Acknowledgments
†This work was supported by the National Institutes of Health, NIGMS Grant # GM074249, and # GM079383, NICHD Grant # HD060559, and # NCRR 1 S10 RR17214-01 from the National Center for Research Resources, NSF grant #MCA017S027, and the University of Utah Center for High Performance Computing.
Abbreviations
- SERM
selective estrogen receptor modulator
- GSH
glutathione
- LC/MS
liquid chromatography-mass spectrometry
- ESI
electrospray ionization
- CID
collision-induced dissociation
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DLPC
1,2-dilauryl-sn-glycero-3-phophocholine
- DLPS
1,2-diacyl-sn-glycero-phospho-L-serine
References
- 1.Rendic S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug metabolism reviews. 2002;34:83–448. doi: 10.1081/dmr-120001392. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chemical research in toxicology. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 3.Guengerich FP. Uncommon P450-catalyzed reactions. Current drug metabolism. 2001;2:93–115. doi: 10.2174/1389200013338694. [DOI] [PubMed] [Google Scholar]
- 4.Yost GS. Bioactivation of toxicants by cytochrome P450-mediated dehydrogenation mechanisms. Advances in experimental medicine and biology. 2001;500:53–62. doi: 10.1007/978-1-4615-0667-6_6. [DOI] [PubMed] [Google Scholar]
- 5.Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chemical research in toxicology. 2004;17:3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- 6.Regal KA, Laws GM, Yuan C, Yost GS, Skiles GL. Detection and characterization of DNA adducts of 3-methylindole. Chemical research in toxicology. 2001;14:1014–1024. doi: 10.1021/tx0100237. [DOI] [PubMed] [Google Scholar]
- 7.Guengerich FP, Kim DH. Enzymatic oxidation of ethyl carbamate to vinyl carbamate and its role as an intermediate in the formation of 1,N6-ethenoadenosine. Chemical research in toxicology. 1991;4:413–421. doi: 10.1021/tx00022a003. [DOI] [PubMed] [Google Scholar]
- 8.Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug metabolism reviews. 2002;34:17–35. doi: 10.1081/dmr-120001387. [DOI] [PubMed] [Google Scholar]
- 9.Kumar D, De Visser SP, Shaik S. Oxygen economy of cytochrome P450: What is the origin of the mixed functionality as a dehydrogenase-oxidase enzyme compared with its normal function? Journal of the American Chemical Society. 2004;126:5072–5073. doi: 10.1021/ja0318737. [DOI] [PubMed] [Google Scholar]
- 10.Kartha JS, Skordos KW, Sun H, Hall C, Easterwood LM, Reilly CA, Johnson EF, Yost GS. Single mutations change CYP2F3 from a dehydrogenase of 3-methylindole to an oxygenase. Biochemistry. 2008;47:9756–9770. doi: 10.1021/bi8005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoch GA, Attias R, Le Ret M, Werck-Reichhart D. Key substrate recognition residues in the active site of a plant cytochrome P450, CYP73A1. Homology guided site-directed mutagenesis. European journal of biochemistry / FEBS. 2003;270:3684–3695. doi: 10.1046/j.1432-1033.2003.03739.x. [DOI] [PubMed] [Google Scholar]
- 12.Szklarz GD, Halpert JR. Use of homology modeling in conjunction with site-directed mutagenesis for analysis of structure-function relationships of mammalian cytochromes P450. Life sciences. 1997;61:2507–2520. doi: 10.1016/s0024-3205(97)00717-0. [DOI] [PubMed] [Google Scholar]
- 13.Lewis DF, Lake BG, Dickins M, Goldfarb PS. Homology modelling of CYP2A6 based on the CYP2C5 crystallographic template: Enzyme-substrate interactions and QSARs for binding affinity and inhibition. Toxicol In Vitro. 2003;17:179–190. doi: 10.1016/s0887-2333(02)00132-7. [DOI] [PubMed] [Google Scholar]
- 14.Lewis DF, Lake BG, Dickins M, Goldfarb PS. Homology modelling of CYP3A4 from the CYP2C5 crystallographic template: Analysis of typical CYP3A4 substrate interactions. Xenobiotica; the fate of foreign compounds in biological systems. 2004;34:549–569. doi: 10.1080/00498250410001691325. [DOI] [PubMed] [Google Scholar]
- 15.Sun H, Moore C, Dansette PM, Kumar S, Halpert JR, Yost GS. Dehydrogenation of the indoline-containing drug 4-chloro-N-(2-methyl-1-indolinyl)-3-sulfamoylbenzamide (indapamide) by CYP3A4: Correlation with in silico predictions. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:672–684. doi: 10.1124/dmd.108.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. Journal of medicinal chemistry. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 17.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 18.Oda A, Y N, Hirono S. New AMBER force field parameters of heme iron for cytrochrome P450s determined by quantum chemical calculations of simplified models. J Comp Chem. 2005;26:818–826. doi: 10.1002/jcc.20221. [DOI] [PubMed] [Google Scholar]
- 19.Chen Q, Ngui JS, Doss GA, Wang RW, Cai X, DiNinno FP, Blizzard TA, Hammond ML, Stearns RA, Evans DC, Baillie TA, Tang W. Cytochrome P450 3A4-mediated bioactivation of raloxifene: Irreversible enzyme inhibition and thiol adduct formation. Chemical research in toxicology. 2002;15:907–914. doi: 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- 20.Yu L, Liu H, Li W, Zhang F, Luckie C, van Breemen RB, Thatcher GR, Bolton JL. Oxidation of raloxifene to quinoids: Potential toxic pathways via a diquinone methide and o-quinones. Chemical research in toxicology. 2004;17:879–888. doi: 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]
- 21.Baer BR, Wienkers LC, Rock DA. Time-dependent inactivation of P450 3A4 by raloxifene: Identification of Cys239 as the site of apoprotein alkylation. Chemical research in toxicology. 2007;20:954–964. doi: 10.1021/tx700037e. [DOI] [PubMed] [Google Scholar]
- 22.Yukinaga H, Takami T, Shioyama SH, Tozuka Z, Masumoto H, Okazaki O, Sudo K. Identification of cytochrome P450 3A4 modification site with reactive metabolite using linear ion trap-Fourier transform mass spectrometry. Chemical research in toxicology. 2007;20:1373–1378. doi: 10.1021/tx700165q. [DOI] [PubMed] [Google Scholar]
- 23.Moore CD, Reilly CA, Yost GS. CYP3A4-Mediated oxygenation versus dehydrogenation of raloxifene. Biochemistry. 2010;49:4466–4475. doi: 10.1021/bi902213r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science (New York, N Y. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 25.Morris GM, G D, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 26.Domanski TL, Liu J, Harlow GR, Halpert JR. Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: Role in steroid hydroxylase activity and alpha-naphthoflavone stimulation. Archives of biochemistry and biophysics. 1998;350:223–232. doi: 10.1006/abbi.1997.0525. [DOI] [PubMed] [Google Scholar]
- 27.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. The Journal of biological chemistry. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 28.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I Evidence for its hemoprotein nature. The Journal of biological chemistry. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 29.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. The Journal of biological chemistry. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 30.Anzenbacher P, Hudecek J. Differences in flexibility of active sites of cytochromes P450 probed by resonance Raman and UV-Vis absorption spectroscopy. Journal of inorganic biochemistry. 2001;87:209–213. doi: 10.1016/s0162-0134(01)00389-0. [DOI] [PubMed] [Google Scholar]
- 31.Anzenbacherova E, Bec N, Anzenbacher P, Hudecek J, Soucek P, Jung C, Munro AW, Lange R. Flexibility and stability of the structure of cytochromes P450 3A4 and BM-3. European journal of biochemistry / FEBS. 2000;267:2916–2920. doi: 10.1046/j.1432-1327.2000.01305.x. [DOI] [PubMed] [Google Scholar]
- 32.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chemical reviews. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 33.Park H, Lee S, Suh J. Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. Journal of the American Chemical Society. 2005;127:13634–13642. doi: 10.1021/ja053809q. [DOI] [PubMed] [Google Scholar]
- 34.Khan KK, He YQ, Domanski TL, Halpert JR. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: An evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Molecular pharmacology. 2002;61:495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- 35.Stevens JC, Domanski TL, Harlow GR, White RB, Orton E, Halpert JR. Use of the steroid derivative RPR 106541 in combination with site-directed mutagenesis for enhanced cytochrome P-450 3A4 structure/function analysis. The Journal of pharmacology and experimental therapeutics. 1999;290:594–602. [PubMed] [Google Scholar]
- 36.Davydov DR, Davydova NY, Tsalkova TN, Halpert JR. Effect of glutathione on homo- and heterotropic cooperativity in cytochrome P450 3A4. Arch Biochem Biophys. 2008;471:134–145. doi: 10.1016/j.abb.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]