

Abstract
The myriad functions of lipids as signalling molecules is one of the most interesting fields in contemporary pharmacology, with a host of compounds recognized as mediators of communication within and between cells. The N-acyl conjugates of amino acids and neurotransmitters (NAANs) have recently come to prominence because of their potential roles in the nervous system, vasculature and the immune system. NAAN are compounds such as glycine, GABA or dopamine conjugated with long chain fatty acids. More than 70 endogenous NAAN have been reported although their physiological role remains uncertain, with various NAAN interacting with a low affinity at G protein coupled receptors (GPCR) and ion channels. Regardless of their potential physiological function, NAAN are of great interest to pharmacologists because of their potential as flexible tools to probe new sites on GPCRs, transporters and ion channels. NAANs are amphipathic molecules, with a wide variety of potential fatty acid and headgroup moieties, a combination which provides a rich source of potential ligands engaging novel binding sites and mechanisms for modulation of membrane proteins such as GPCRs, ion channels and transporters. The unique actions of subsets of NAAN on voltage-gated calcium channels and glycine transporters indicate that the wide variety of NAAN may provide a readily exploitable resource for defining new pharmacological targets. Investigation of the physiological roles and pharmacological potential of these simple lipid conjugates is in its infancy, and we believe that there is much to be learnt from their careful study.
Keywords: molecular pharmacology, neuropharmacology, calcium channels, cannabinoids, glycine, other transporters, transient receptor potential channels, analgesics drugs, anti-inflammatory drugs, pain
Introduction
The myriad functions of lipids as signalling molecules has emerged as one of the most interesting fields in contemporary pharmacology, with a host of compounds being recognized as mediators of communication within and between cells (Piomelli et al., 2007). Much attention has been focussed on the synthesis, catabolism and cellular effects of lipid ligands for the cannabinoid CB1 G protein coupled receptor (GPCR). The first endogenous ligand identified for this receptor, N-arachidonoyl ethanolamide (anandamide, Devane et al., 1992), represents the prototype for the N-acyl ethanolamides, a large family of lipid signalling molecules which vary in hydrocarbon chain length and saturation (Di Marzo et al., 1994). Recently, another family of molecules has been identified in mammals which differ from anandamide in that rather than ethanolamide, compounds such as glycine, GABA or dopamine are linked to fatty acids to form N-acyl amino acid/neurotransmitter (NAAN) conjugates. More than 70 endogenous NAAN have been reported from mammals (Huang et al., 2001, 2002; Chu et al., 2003; Milman et al., 2006; Saghatelian et al., 2004; 2006; Rimmerman et al., 2008; Bradshaw et al., 2009b; Tan et al., 2010). The structures of a subset of these compounds are presented in Figure 1. Most NAAN are differentially distributed in the body, several undergo hormonal regulation of their levels in the brain (Bradshaw et al., 2006) and they have a range of biological activities in the nervous system, vasculature and immune system although the functional effects of many recently identified NAAN remain unexplored. In addition to their physiological roles, NAAN are interesting because of their potential as flexible pharmacological tools to probe new sites on GPCRs, transporters and ion channels (Table 1). NAAN effects have been studied for some time in invertebrates and plants; we will restrict our discussion to NAAN that have been found in mammals.
Figure 1.
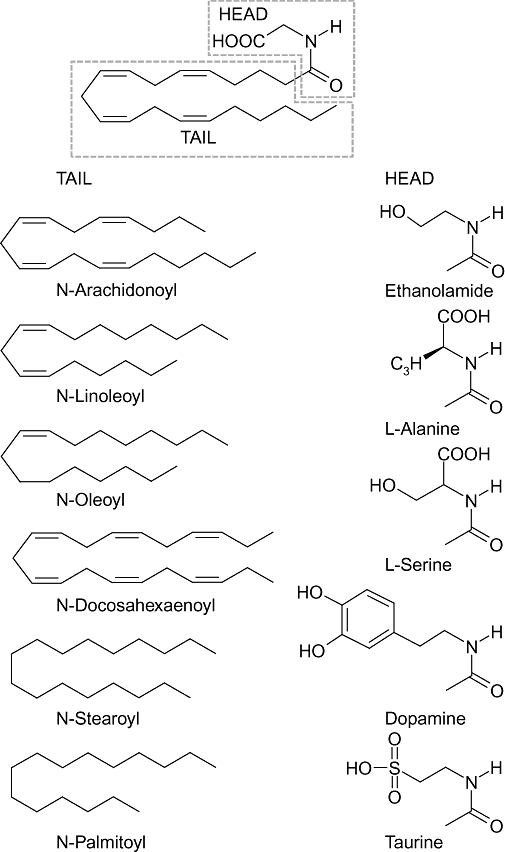
Structures of N-acyl amino acid/neurotransmitter (NAAN). The prototypical NAAN, N-arachidonoyl glycine (NA-Gly) is shown with the tail and head groups circled. The various NAAN differ in the length and degree of saturation of the hydrophobic acyl tail group. NA-Gly is closely related to anandamide, with the only difference being that NA-Gly has a carboxyl group compared with a hydroxyl group on anandamide. Other amino acid or neurotransmitter head groups are also shown.
Table 1.
A summary of the actions of arachidonic acid, anandamide and endogenous NAAN at well defined cellular effectors. The value in parentheses represents the approximate pEC50 for the indicated effect. For details of assays and criteria for lack of activity, see cited references. Details of actions of NAAN at presently undefined receptors can be found in the main text, as can reports of the activity of a single NAAN at a given molecule. Where the activity of a given NAAN at a given effector has been assessed by several groups, initial reports, papers where several compounds have been compared and conflicting studies have been cited
| G protein coupled receptors | Calcium channels | TRP channels | Glycine transporters | FAAH | |
|---|---|---|---|---|---|
| Arachidonic acid | Inhibits T-type (5.5) Chemin et al., 2001, 2007Inhibits/Potentiates N-type (5.5) Barrett et al., 2001; Liu et al., 2001 | Inhibits GLYT1 (5.5) Inactive at GLYT2 Wiles et al., 2006 | |||
| Anandamide | CB1 agonist CB2 agonist GPR55 agonist Ross, 2009 | Inhibits T-type (6) Chemin et al., 2001Inhibits high voltage activated (<5) Guo and Ikeda, 2004 | TRPV1 agonist (5.5) Huang et al., 2002TRPM8 antagonist (6.3) De Petrocellis et al., 2007 | Stimulates GLYT1 (4.7) Inactive at GLYT2 Wiles et al., 2006 | Substrate |
| NA-Gly | GPR18 Agonist (7.7) Kohno et al., 2006Inactive at GPR18 Yin et al., 2009GPR92/LPA5 receptor partial agonist Oh et al., 2008 (5.3) LPA2, LPA5 receptor agonist (weak) LPA1, LPA3, LPA4 antagonist (6.2) Williams et al., 2009 | Inhibits T-type (5, 6) Ross et al., 2009,Barbara et al., 2009Potentiates N-type (5) Guo et al., 2008 | Inactive at TRPV1 De Petrocellis et al., 2000,Ross et al., 2009,Barbara et al., 2009 | Inhibits GLYT2 (5.3) Inactive at GLTY1 Inactive at GAT1 Wiles et al., 2006 | Inhibits (5.3) Huang et al., 2001 |
| NA-Ala | Potentiates N-type (5) Guo et al., 2008Inhibits T-type (6) Barbara et al., 2009 | Inhibits GLYT2 (5.5) Inactive at GLTY1 Inhibits GAT1 (4.7) Wiles et al., 2006 | Inhibits (4.5) Cascio et al., 2004 | ||
| NA-GABA | GPR92/LPA5 receptor partial agonist Oh et al., 2008 (5.3) | Inhibits T-type (6) Barbara et al., 2009 | Inhibits GLYT2 (5.5) Inactive at GLTY1 Inactive at GAT1 Wiles et al., 2006 | ||
| NA-DA | CB1 agonist (6.5) CB2 ligand (5) Bisogno et al., 2000Inactive at GPR119 Chu et al., 2010 | Inhibits T-type (6.3) Ross et al., 2009Inactive at N-type (5) Guo et al., 2008 | TRPV1 agonist (6.1) De Petrocellis et al., 2000,Huang et al., 2002TRPM8 antagonist (6) De Petrocellis et al., 2007 | Inhibits (4.7) Bisogno et al., 2000 | |
| OL-DA | CB1 ligand (6) Chu et al., 2003GPR119 agonist (5.5) Chu et al., 2010 | Inhibits T-type Ross et al., 2009 | TRPV1 agonist Chu et al., 2003 | Inactive Chu et al., 2003 | |
| NA-Ser | Inactive at CB1 Inactive at CB2 Milman et al., 2006 | Inhibits T-type (6) Barbara et al., 2009Potentiates N-type (5) Guo et al., 2008 | Inactive at TRPV1 Milman et al., 2006 | ||
| NA-Tau | Inhibits T-type (6) Barbara et al., 2009 | TRPV1 agonist TRPV4 agonist Saghatelian et al., 2006 |
NAAN, N-acyl amino acid/neurotransmitter.
Physical properties of NAAN
The physical properties of NAAN need to be kept in mind when considering their functional properties. NAAN are amphiphiles, consisting of a hydrocarbon tail and a polar head group (Figure 1). The head group may be charged (e.g. N-arachidonoyl glycine, NA-Gly) or neutral [e.g. N-arachidonoyl dopamine (NA-DA)]. The hydrocarbon tail of NAANs allows them to intercalate into cell membranes, potentially altering the biophysical properties of the membranes, much as detergents do, and like detergents, NAAN could affect the functional properties membrane proteins this way. Headgroup charge means some NAAN may also potentially affect ion channel function through surface charge screening effects at high concentration, and it will also affect their propensity to penetrate into membranes; however, neither detergent or charge effects of NAAN has been explicitly examined. The best studied NAAN effects are on transient receptor potential vanilloid 1 receptors (TRPV1), the glycine transporter GLYT2 and T-type calcium channels (ICa) and these effects are inconsistent with simple perturbation of membrane properties. In the cases of GLYT2 and T-type ICa the effects of the NAAN point to the existence of novel sites on channels and transporters that have the potential to be manipulated by pharmacological means (Figure 2).
Figure 2.
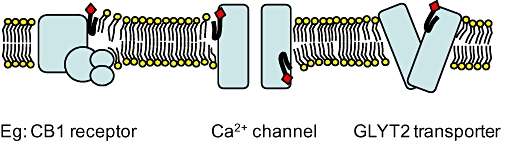
Potential sites of action for N-acyl amino acid/neurotransmitter (NAAN) on receptors, channels and transporters. A variety of sites has been proposed for NAAN binding to membrane proteins, some possible ones are illustrated here. Very little is known about NAAN interactions with G protein coupled receptors (GPCRs), but some possibilities are that the NAAN may disrupt lipid packing at the GPCR-lipid interface and alter the dynamics of protein signaling. The head group may provide specificity, while the lipid tail may alter membrane dynamics. Both inhibitory and stimulatory binding sites have been proposed for Ca2+ channels. NAAN may diffuse through the membrane to interact with intracellular binding sites of ion channels and alter the kinetics of channel function. NAAN may bind to extracellular recognition sites of transporters and alter the conformational changes associated with the transport process.
Additional important considerations in distinguishing between specific interactions between NAAN and potential targets and non-specific membrane effects are the concentration the NAANs produced and the accessibility of the compounds to their potential site of action. While amounts of NAAN present in a variety of tissues has been reported by several investigators (see next), no attempt has been made to estimate the free concentrations of the compounds, and as yet there is no indication where NAAN are found within cells, or whether they are released from cell membranes. One potential difficultly in accurately estimating effective concentrations of NAAN both in vivo and in in vitro experiments is that these compounds will form micelles and have an inherent propensity to be incorporated in lipid membranes. The critical micelle concentration for NAANs is likely to be in the range of 30–100 µM, making any observed effects in this concentration range difficult to interpret. In the following review, we will briefly discuss how NAAN are produced, describe the apparent physiological actions of a number of NAAN and consider how they may interact with and manipulate receptors, channels and transporters.
Biosynthesis and metabolism of NAAN
The mechanisms responsible for the formation and degradation of NAAN are not completely defined and a full review of these areas is beyond the scope of this article. Two major types of biosynthetic route have been proposed for NAAN. The first is the conjugation of the amino acid/neurotransmitter with arachidonic acid or arachidonoyl coenzyme A, the second is the sequential modification of a precursor fatty acid conjugate to form the final NAAN. Examples of each route have been described for the formation of N-acyl glycines (Huang et al., 2001; Merkler et al., 2004; McCue et al., 2008; Aneetha et al., 2009) and been suggested for NA-DA (Huang et al., 2002), although it seems likely that NA-DA is formed predominantly through FAAH-mediated condensation (Hu et al., 2009). There is some evidence for differential cellular distribution of the distinct pathways for NA-Gly synthesis in different cell types (Bradshaw et al., 2009a) and the physiological regulation of NAAN synthesis is a topic of intense investigation.
The metabolism of NAAN is likely to be more complex, with at least three major types of metabolic pathways identified. The first of these is hydrolysis by fatty acid amide hydrolase (FAAH) to produce the free fatty acid and neurotransmitter/amino acid. FAAH is the major enzyme responsible for terminating the action of anandamide (Cravatt et al., 1996), and it may serve as a primary source of arachidonic acid for the biosynthesis of some NAAN (Bradshaw et al., 2009a). The other two major types of metabolism are modification of the acyl moiety or the NAAN headgroup. Many enzymes modify fatty acid groups, while each amino acid/neurotransmitter potentially has its own series of metabolic enzymes. There is significant potential for NAAN interaction with metabolic enzymes to contribute significantly to the biological activity of the NAAN, either via enzyme inhibition or a change in the target profile of the metabolite.
N-acyl glycine
N-arachidonoyl glycine is the carboxylic acid congener of anandamide and was originally synthesized as part of a study of anandamide structure-activity relationships at the CB1 receptor (Sheskin et al., 1997). It is the simplest N-arachidonoyl amino acid and was the first identified in mammals (Huang et al., 2001). Endogenous NA-Gly is not uniformly distributed in the body, with highest levels found in the spinal cord and small intestine (∼140 pmol g-1 dry tissue weight), lower levels in brain, kidney and skin (∼80–100 pmol g−1) and very low levels in the heart (<5 pmol g−1, Huang et al., 2001). N-oleoyl glycine (NO-Gly) was postulated as an intermediate in the synthesis of oleamide (Merkler et al., 2004) and is most abundant in skin and lung. N-palmitoyl glycine (NP-Gly), N-stearoyl, N-linoleoyl and N-docosahexaenoyl glycine are most abundant in skin and/or lung (Rimmerman et al., 2008; Bradshaw et al., 2009b), with levels up to 10-fold higher than that of NA-Gly, and NP-Gly is also found in appreciable amounts in spinal cord and intestine.
NA-Gly metabolism potentially provides links with its biological activities. NA-Gly is metabolized by COX-2 to prostaglandin H2-glycine (PGH2-Gly; Burstein et al., 2000; Prusakiewicz et al., 2002) and by FAAH to arachidonic acid and glycine. The catalytic efficiency of COX-2 for the generation of PGH2-Gly is only 10% of that for the production of PGH2 from arachidonic acid while FAAH hydrolizes NA-Gly much more slowly than its principal substrate, anandamide. Thus, NA-Gly may be thought of as an endogenous inhibitor of both COX-2 and FAAH (Prusakiewicz et al., 2002; Cascio et al., 2004), although the biological significance if this remains to be demonstrated.
In vivo effects
Anti-nociception is the most intensively studied biological effect of NA-Gly. Systemic or local administration of NA-Gly reduces responses to a noxious thermal stimulus in mouse (Burstein et al., 2000; Barbara et al., 2009) and local administration of NA-Gly into mouse paw inhibits nociceptive behaviours produced by co-injection of formalin (Huang et al., 2001). Intrathecal administration of NA-Gly partially reverses mechanical allodynia and thermal hyperalgesia produced by paw inflammation (Succar et al., 2007) and also partially reverses mechanical allodynia produced by chronic spinal nerve ligation (Vuong et al., 2008).
The mechanism underlying the thermal antinociceptive effect of locally applied NA-Gly is likely to involve inhibition of T-type ICa (Barbara et al., 2009; Ross et al., 2009), specifically CaV3.2 (Barbara et al., 2009). When injected into the paw, NA-Gly increases the paw withdrawal latency to a noxious thermal stimulus, and this effect is abolished in CaV3.2 knockout mice (Barbara et al., 2009). The mechanism(s) responsible for the antinociceptive effects of NAGly administered via different routes or in other animal pain models remain unknown. For example, while NA-Gly can inhibit FAAH and enhance anandamide levels when administered systemically (Burstein et al., 2002), the anti-nociceptive effects of intrathecally administered NA-Gly were not blocked by either CB1 or CB2 receptor antagonists (Succar et al., 2007; Vuong et al., 2008), ruling out direct or indirect roles of these receptors. NP-Gly also has in vivo actions consistent with an anti-nociceptive activity; injection of NP-Gly into in the paw of anaesthetized rats inhibits the activation of dorsal horn neurons by noxious thermal stimuli applied to the paw (Rimmerman et al., 2008).
NA-Gly has also been tested in a variety of in vitro and in vivo assays of inflammation and immune cell function. NA-Gly reduces proliferation of stimulated human T lymphocytes and modestly suppresses interleukin1b release from mononuclear cells (Burstein et al., 2000). Several glycine series NAANs, including NA-Gly and NO-Gly (but not NP-Gly), also reduce proliferation of macrophage RAW 264.67 cells and have anti-inflammatory effects in a range of in vivo assays. The mechanisms underlying these effects remain to be determined (Burstein et al., 2007) but possible sites of action include COX-2 (Prusakiewicz et al., 2002) and peroxisome proliferator-activated receptors, a recently described target of endo- and phyto-cannabinoids (O'Sullivan, 2007).
N-arachidonoyl GABA
N-arachidonoyl GABA (NA-GABA) was also synthesized as an anandamide congener (Burstein et al., 2000) before its isolation from brain tissue (Huang et al., 2001). NA-GABA levels are generally similar to those of NA-Gly, although they differ about twofold between brain regions and modestly across the oestrus cycle in rats (Bradshaw et al., 2006). Unlike NA-Gly, no NA-GABA is found in the pituitary (Bradshaw et al., 2006). NA-GABA has very modest anti-nociceptive effect after systemic administration (Burstein et al., 2000) but lacks anti-nociceptive activity in a rat inflammatory pain model after intrathecal administration (Succar et al., 2007). The NA-GABA analog NA-GABA-OH is an effective inhibitor of thermal nociception when administered locally into mouse paw (Barbara et al., 2009).
N-arachidonoyl alanine
N-arachidonoyl alanine (NA-Ala) was isolated from brain tissue along with NA-Gly and NA-GABA (Huang et al., 2001). Like NA-GABA, NA-Ala did not produce anti-nociception in a model of inflammatory pain in rat (Succar et al., 2007), and is a very weak inhibitor of FAAH (Cascio et al., 2004). N-acyl alanines are active in the assays of RAW 264.7 cell function and a subset of the in vivo assays of inflammation described in Burstein et al. (2007) but the mechanisms underlying these effects are unknown. The reported cellular actions of NA-Ala include enhancement of N-type ICa (Guo et al., 2008), inhibition of T-type ICa (Barbara et al., 2009) and inhibition of the glycine transporter, GLYT2 (Wiles et al., 2006). These effects are similar to those of NA-Gly and N-arachidonoyl serine (NA-Ser), discussed below.
N-acyl dopamine
N-acyl dopamine compounds were first described as potent inhibitors of 5-lipoxygenase (Tseng et al., 1992), while N-arachidonoyl dopamine was synthesized as a possible inhibitor of FAAH (Bisogno et al., 2000). NA-DA was isolated from bovine brain (Huang et al., 2002) and oleoyl-, palmitoyl- and stearoyl-dopamine (NO-DA, NP-DA, NS-DA) reported shortly afterwards (Chu et al., 2003). NA-DA has a restricted distribution in the brain, with levels highest in the striatum (∼5 pmol g−1 wet tissue weight) and very low elsewhere (Huang et al., 2002; Bradshaw et al., 2006). NA-DA has not been identified outside the brain.
The actions of NA-DA in vivo are strikingly dependent on the route of administration. Systemic injection of NA-DA had anandamide-like activity in mice, producing the classic tetrad of behavioural signs characteristic of cannabinoid agonists such as increased immobility in the ring test, decreased body temperature, reduced locomotor activity and delayed response to a thermal stimulus (Bisogno et al., 2000; Bezuglov et al., 2001). However, the involvement of cannabinoid receptors in these effects was not explicitly addressed. Systemic NA-DA also reduces opioid-induced emesis in ferrets via mechanisms broadly consistent with its actions at both TRPV1 and CB1 receptors (Sharkey et al., 2007), but additionally reduces spontaneous motor activity in via mechanisms partially independent of TRPV1 or CB1 receptors (Sharkey et al., 2007).
By contrast, when NA-DA or NO-DA is administered in the periphery, they generally behave like the prototypic TRPV1 agonist capsaicin. Injection of either compound into the hindpaw of rats produces thermal allodynia (Huang et al., 2002; Chu et al., 2003) and injection of NO-DA is accompanied by spontaneous paw lifting and licking. These behaviours are blocked by a TRPV1 antagonist (Chu et al., 2003). Similar results were reported in mouse, where the sensitizing effects of NO-DA to heat were strongly reduced in TRPV1 knockout animals (Szolcsanyi et al., 2004). Consistent with the effects observed in rodents, both NA-DA and NO-DA produced thermal allodynia after topical administration to the tail of monkeys (Butelman et al., 2003, 2004).
N-arachidonoyl serine
NA-Ser was also originally synthesized as an analog of anandamide (Sheskin et al., 1997) and was isolated from bovine brain by Milman et al. (2006). The effect of systemic or central nervous system administration of NA-Ser has not been reported. NA-Ser has negligible affinity for CB1 or CB2 receptors (Sheskin et al., 1997; Milman et al., 2006) but a complex in vitro pharmacology is becoming apparent, as discussed next.
N-acyl taurine
N-acyl taurine (NAc-Tau) conjugates were discovered by comparing the brain metabonome of FAAH knockout mice with their wild-type controls (Saghatelian et al., 2004). The concentrations of the previously unknown acyl-taurine compounds were elevated by 10 to 100-fold in the FAAH knockouts, increases at least as great as those seen with the N-acyl ethanolamides (Saghatelian et al., 2004, 2006). NAc-Taus were subsequently found in liver and kidney, but the particular species found in the periphery differed strikingly from brain (Saghatelian et al., 2006). The predominant species in liver and kidney were polyunsaturated compounds, including N-arachidonoyl taurine (NA-Tau, ∼20 pmol g−1 liver, ∼160 pmol g−1 kidney), while long chain (C22-C24) saturated and mono-unsaturated species were the most common in brain (up to 2 nmol g−1, Saghatelian et al., 2006). The function(s) of the NAc-Tau remain largely unexplored, although NA-Tau is a weak activator of several transient receptor potential (TRP) channels and an inhibitor of T-type ICa (Barbara et al., 2009).
Actions of NAAN at GPCR
Anandamide is an agonist for the GPCRs CB1, CB2 and GPR55 (Ross, 2009), with many of its most important biological effects mediated via the CB1 receptor. Some NAAN may be physiologically relevant GPCR agonists as well, but current data are sparse and inconsistent. Identifying endogenous ligands for poorly characterized GPCR can be fraught, and lipid-related compounds seem particularly prone to cause discordant results (see Seuwen et al., 2006; Ross, 2009). NAANs are not known to activate GPR55, but the complexities underlying the unresolved pharmacology of this receptor should be kept in mind when attempting to reconcile the possible GPCR activities of NAAN (Ross, 2009). Importantly, although there are a significant number of orphan GPCR (∼140, see Harmar et al., 2009), there are also at least 30 receptors known to be activated by lipophilic molecules such as lysophosphotidylinositol derivatives, sphingosine-1-phosphate, free fatty acids, fatty acid ethanolamides, eicosanoids and related molecules (see Alexander et al., 2009). Some of these GPCRs are affected by NAAN, albeit with modest potency (Williams et al., 2009).
Cannabinoid CB1 and CB2 receptors
NA-DA is the only NAAN that has been directly shown to be a CB receptor agonist. However, the affinity of NA-DA and other N-acyl dopamine compounds for CB1 receptors is modest, and there is little information about their relative potency and efficacy compared with other cannabinoid receptor agonists. In binding assays at CB1 receptors, the Ki for NA-DA is 250–500 nM (Bisogno et al., 2000; Bezuglov et al., 2001; Chu et al., 2003) while OL-DA has lower affinity (Ki 1.5 µM, Chu et al., 2003). NA-DA stimulates calcium mobilization in neuroblastoma cells in a manner consistent with activation of CB1 receptors (Bisogno et al., 2000) and inhibits GABA release onto dopaminergic neurons of the substantia nigra via CB1 receptor activation (Marinelli et al., 2007). The signalling of NA-DA and other acyl dopamine ligands has not been studied at recombinant CB receptors, and it is not known whether OL-DA is a CB1 agonist or antagonist. NA-Gly has been suggested to act in a CB2 receptor-dependent manner (Sipe et al., 2005), but a direct interaction has not been demonstrated.
GPR18 and GPR92
NA-Gly has been reported to be an agonist for GPR18 (Kohno et al., 2006) and GPR92 (Oh et al., 2008). GPR18 is expressed in the testis and components of the immune system while GPR92 is found more widely, including in brain and sensory neurons (Oh et al., 2008). NA-Gly potently (EC50 20 nM) inhibited adenylyl cyclase activity in GPR18-transfected Chinese hamster ovary (CHO) cells and enhanced elevations of intracellular calcium in response to NA-Gly are observed in several cell lines following GPR18 transfection (Kohno et al., 2006). However, a recent study using different bioassays to that of Kohno et al. (2006) found no activity for NA-Gly at GPR18 (Yin et al., 2009). NA-Gly effects on native GPR18 have not been reported.
Several studies that have investigated recombinant GPR92/LPA5 receptor pharmacology have found relatively weak activation of the receptor by NA-Gly at concentrations between 1 and 30 µM. (Oh et al., 2008; Williams et al., 2009; Yin et al., 2009). Maximal NA-Gly effects were much less than those produced by other endogenous agonists lysophosphatidic acid and farnesyl pyrophosphate. Some evidence for NA-Gly activation of native GPR92 was provided by the observation that elevations of intracellular calcium in sensory neurons by NA-Gly were strongly reduced by siRNA targeting GPR92 (Oh et al., 2008). NA-Gly was reported to be a weak antagonist of signalling through LPA1, LPA3 and LPA4 receptors and a weak agonist at LPA2 receptors (Williams et al., 2009). NP-Gly stimulates Ca2+ influx in F-11 neuroblastoma hybrid cells by a mechanism partially sensitive to the inhibitor of G protein activation pertussis toxin (PTX) and this effect was not mimicked by NA-Gly (Rimmerman et al., 2008), implying an action at a site distinct from GPR18 or GPR92.
GPR119
Oleoyl dopamine is one of a group of lipids which activate GPR119, a Gs coupled receptor found in pancreatic islet cells and gut (Chu et al., 2010). OL-DA promoted insulin secretion from pancreatic cell lines in vitro and improved glucose tolerance after oral administration in wild-type mice but not those with a deletion of GPR119 (Chu et al., 2010). Intriguingly, NA-DA was inactive at GPR119, as was N-oleoyl tyrosine, suggesting that ligand recognition by the receptor involved elements of both the fatty acid moiety and head group of the NAAN (Chu et al., 2010).
Unidentified putative GPCR
A putative endothelial cannabinoid receptor, the Abnormal-cannabidiol receptor (Abn-CBD receptor), mediates several effects including an endothelium-dependent relaxation of some vascular beds (Jarai et al., 1999) and inhibition of N-formyl-methionyl-leucyl-phenylalanine-induced migration of human neutrophils (McHugh et al., 2008). Anandamide, Abn-CBD and O-1602 are agonists at this receptor while cannabidiol and the related molecule O-1918 are antagonists. NA-DA, NA-Gly, NA-Ser and NA-GABA may act at least partially through the Abn-CBD receptor to produce relaxation of mesenteric blood vessels (O'Sullivan et al., 2004; Parmar and Ho, 2010) while NA-DA also inhibits neutrophil migration (McHugh et al., 2008). NA-Ser mimics the biochemical effects of Abn-CBD receptor activation in human umbilical vein endothelial fibroblasts, including potentiation of large conductance calcium activated potassium channel (BKCa) activity and stimulation of p42/44 mitogen activated protein kinase and protein kinase B signalling (Milman et al., 2006). Intriguingly, however, in the assay of neutrophil migration NA-Ser was reported to be an Abn-CBD receptor antagonist (McHugh et al., 2008). Comprehensive studies comparing the potency and efficacy of Abn-CBD receptor agonists have not been reported, so whether NA-Ser is a partial agonist or perhaps couples to only a subset of possible effector pathways is unknown. The similar pharmacological profiles of GPR55 and the Abn-CBD receptor raises the possibility that some of the effects of NA-DA or NA-Ser attributed to activation of the Abn-CBD receptor are mediated by GPR55, although there is strong evidence that GPR55 and the Abn-CBD receptor are distinct molecular entities, at least in blood vessels (Johns et al., 2007).
There may be other GPCR-linked sites of action for NA-DA and NA-Ser on blood vessels. Both compounds produce relaxation of rat aorta that is PTX-sensitive but only partially blocked by endothelium removal. The effects are similar to those produced through activation the endothelial cannabinoid receptor, but in this vessel the effects of anandamide, Abn-CBD and NA-Ser are largely insensitive to the antagonist O-1918 (O'Sullivan et al., 2005; Milman et al., 2006; Herradon et al., 2007). The sensitivity of NA-DA vasorelaxation in aorta to O-1918 has not been tested, but the data point to the existence of a GPCR activated by NA-DA and NA-Ser that is distinct from the Abn-CBD receptor.
Actions of NAAN at transient receptor potential channels
Transient receptor potential channels are a large family of cation channels whose various members can be activated by an enormous range of ligands and environmental stimuli. Many of the ligands for TRP channels are lipophilic and include arachidonic acid-derived molecules such as O-acyl glycerol, epoxyeicosatrienoic acids and anandamide.
NA-DA, NO-DA and NA-Tau are agonists at TRPV1 (Huang et al., 2002; Saghatelian et al., 2006), while NA-Gly is not (Huang et al., 2001; Ross et al., 2009). The reported efficacy and potency of NA-DA and NO-DA at TRPV1 varies enormously, even among investigators using superficially similar assays. NA-DA and NO-DA seem to be partial agonists, with potencies between that of capsaicin and anandamide (Huang et al., 2002; Chu et al., 2003; Szolcsanyi et al., 2004), although no studies have directly compared the potency of NA-DA or NO-DA with capsaicin in activating TRPV1 currents. When maximal responses to NA-DA and capsaicin at human TRPV1 expressed in CHO cells were normalized to responses produced by pH activation of the channel, NA-DA had about 50% of the efficacy of capsaicin (Sutton et al., 2005). An electrophysiological study of NA-DA activation of native capsaicin receptors in embryonic rat sensory neurons suggests a somewhat lower efficacy (Premkumar et al., 2004). NA-Tau activation of TRPV1 is weak, with an EC50 of 30 µM and unknown efficacy (Saghatelian et al., 2006).
NA-DA activation of TRPV1 depends on the same region of the channel crucial for conferring sensitivity to capsaicin and anandamide (Jordt and Julius, 2002; Sutton et al., 2005). Located at the intracellular interface of transmembrane domain 3 and the cytoplasm, this region includes the aromatic Y511 and polar S512 residues, both of which are essential for capsaicin and anandamide gating of the channel. Access to this site appears to depend on a minimal degree of saturation as NO-DA (C18:1) is an agonist while NP-DA (C20:0) is only very weakly active (Chu et al., 2003; De Petrocellis et al., 2004). Anandamide may be a less efficacious TRPV1 agonist than NA-DA, and consistent with this oleamide appears to be a less efficient TRPV1 agonist than NO-DA (Ahern, 2003). The omega hydroxylated metabolites of NA-DA produced by cytochrome P450 activity retain activity at TRPV1, albeit with a lower apparent potency (Rimmerman et al., 2009).
Changing the head group alters NAAN activation of TRPV1. Dopamine conjugates are the most effective activators of TRPV1, and changes in the substituents on the benzene ring of NO-DA have effects that precisely mirror changes made to the benzyl ring of capsaicin (e.g. Walpole et al., 1993). Shielding the 4-hydroxyl group in the aromatic ring of NO-DA with a methyl group abolished TRPV1 agonist activity and produced a weak antagonist (4-MOLDA) but in contrast, N-oleoyl 3-methyl-dopamine (NA-3MeDA or 3-MOLDA), whose aromatic substituents correspond to those of capsaicin, retained agonist activity (Almasi et al., 2008). The potency and efficacy of 3-MOLDA were not compared directly with that of NO-DA (Almasi et al., 2008), but it might be predicted to be a more efficacious agonist than the parent compound. NA-Gly, a carboxylic acid, does not activate TRPV1, but NA-Tau, a sulphonic acid, does albeit weakly. It is not known if NA-Tau interacts with TRPV1 in a manner analogous to NA-DA or capsaicin.
NA-DA or anandamide activation of recombinant TRPV1 is potentiated by co-application of a relatively low concentration (100 nM) of NS-DA or NP-DA, an example of the so-called entourage effect observed with many endocannabinoids (Fowler, 2003; De Petrocellis et al., 2004). The NS-DA or NP-DA concentrations that maximally enhanced TRPV1 activation by NA-DA did not activate TRPV1 by themselves and the stimulatory effect on NA-DA responses was not dose-related over the concentration range examined. The mechanism of the entourage effect remains unknown but both compounds also modestly enhanced pH activation of TRPV1, suggesting that they may be having a general effect on TRPV1 gating, possibly by acting at the unidentified regions of TRPV1 that mediate a residual capsaicin effect on pH gating of the channel after inactivating mutations in the primary capsaicin binding domain (Sutton et al., 2005). The low concentrations at which NS-DA and NP-DA were effective, together with the in vivo evidence for the physiological relevance of entourage effects of acyl ethanolamides suggest that the phenomena warrants further study on TRPV1 and other channels modulated by NA-DA.
NA-Tau has been reported to activate calcium entry through the transient receptor potential vanilloid 4 receptor (TRPV4, Saghatelian et al., 2006) expressed in CHO cells. TRPV4 is a channel potentially involved in sensing osmolarity and mechanical stimuli. NA-Tau was not a potent activator of TRPV4 (EC50 20 µM), but there are few other reported natural agonists. The mechanism underlying the effect of NA-Tau was not determined but its effects were mimicked by arachidonic acid. This is of particular importance because both arachidonic acid and anandamide activate TRPV4 after intracellular metabolism to an epoxyeicosatrienoic acid, raising the possibility that the effects of NA-Tau are also due to metabolites (Watanabe et al., 2003).
In contrast to the NA-DA activation of TRPV1 and NA-Tau activation of TRPV4, agonist activation of recombinant rat TRPM8 is inhibited by NA-DA and anandamide (De Petrocellis et al., 2007). NA-DA antagonized icilin- and menthol-stimulated calcium elevations in TRPM8 expressing HEK 293 cells with a potency of around 1 µM. The effects of NA-DA or other NAAN on native TRPM8 channels or TRPM8 currents remain to be established.
Actions of NAAN at ICa
NAANs have distinctive and contrasting effects on different types of ICa; they can stimulate high voltage activated N-type ICa and inhibit low voltage activated T-type ICa. NA-Ser, NA-Gly and NA-Ala (but not NA-DA) potentiate native N-type ICa in sympathetic neurons (Guo et al., 2008). The stimulation of N-type ICa amplitude was rapid, readily reversible and independent of G-proteins. The basis for the potentiation of ICa was tested for NA-Ser, which was found to produce a hyperpolarizing shift of about −5 mV in the N-type ICa activation curve, meaning that more channels opened following depolarization to relatively negative membrane potentials. The steepness of the ICa activation curve makes this modest shift functionally quite significant. Other studies have reported a similar effect for arachidonic acid (Barrett et al., 2001) and anandamide (Guo and Ikeda, 2004). Interestingly, however, arachidonic acid and anandamide also produce a slower developing inhibition of N-type ICa that was not seen with the NAAN in the study of Guo et al. (2008). Modest inhibition of total sensory neuron HVA ICa has also been reported for NA-Gly and NA-GABA-OH, although the current types affected were not defined (Barbara et al., 2009). It has been suggested that the inhibition of N-type ICa by arachidonic acid is via an intracellular site (Barrett et al., 2001; Liu et al., 2001), and perhaps the NAAN cannot access this site, or are unable to bind to it productively if it is accessible.
In contrast to their effect (or lack of) on N-type ICa, NA-Gly, NA-DA, NA-Ser, NA-Ala, NA-GABA and NA-Tau inhibit recombinant human CaV3 ICa (Barbara et al., 2009; Ross et al., 2009) and where examined, native mouse T-type ICa. This effect is grossly similar to that of anandamide and arachidonic acid (Chemin et al., 2001). NA-DA inhibits recombinant human T-type ICa with EC50 values between 300 nM and 1 µM (Ross et al., 2009), making it one the most potent endogenous inhibitors of these channels identified to date. NO-DA mimics the effects of NA-DA on the cloned human channels but NP-DA is virtually without effect at 10 µM (Ross et al., 2009). NA-Gly was much less potent than NA-DA or anandamide when examined under the same recording conditions (EC50≥10 µM), but when a more depolarized holding potential was utilized (−75 mV vs. −86 mV), NAGly inhibited the channels with EC50 values between 600 nM and 2 µM (Barbara et al., 2009). The reason for this difference in potency arises from the mechanism of action of NAAN and their analogs on CaV3 ICa– each of them produces a profound increase in the degree of steady state channel inactivation at membrane potentials where the channels are closed (Barbara et al., 2009; Ross et al., 2009). Thus, the inhibitory effects of both NA-DA and the NA-GABA analog NA-GABA-OH are almost abolished at holding potentials where CaV3 channels are closed but not inactivated (more negative than −100 mV), but enhanced when cells are held at potentials with significant steady state inactivation (more positive than −80 mV) (Barbara et al., 2009; Ross et al., 2009). Neither NA-DA nor NA-Gly affects the voltage-dependence of channel activation and they have minimal effects on CaV3 kinetics (Ross et al., 2009). This contrasts with the actions of anandamide and arachidonic acid, which also have no affect on the voltage-dependence of activation but which significantly speed channel activation and inactivation from the open state (Chemin et al., 2001, 2007; Ross et al., 2009).
NAANs as probes for the development of a novel pharmacology of ICa
Amphiphilic detergents, arachidonic acid and cholesterol depletion generally have a similar effect on voltage-gated channels – they promote the steady-state inactivation of the channels (Lundbaek, 2008). These effects are thought to result from increased membrane elasticity, which presumably affects the ease with which the intramolecular re-arrangements responsible for different voltage-dependent states of ion channels occur. NA-Gly, NA-DA and anandamide all have a prominent effect on T-type ICa steady-state inactivation, which may reflect a direct effect of these compounds on lipid interactions in the plasma membrane. The differing potencies of NA-DA, NA-Gly and AEA may simply reflect the efficiency with which the structurally distinct molecules affect lipid order, although this has not been directly tested. However, anandamide, NA-DA and other lipophilic cannabinoids such as Δ9-THC and cannabidiol have different rank orders of potency to inhibit different T-type ICa (Ross et al., 2008; 2009;) perhaps implying that rather than simply having a general effect on lipid dynamics these molecules are acting at a specific site on the channels or at a site on the lipid-protein interface that is subtly different among the channels. The fact that NA-DA and NA-Gly do not strongly affect channel kinetics, unlike AEA or arachidonic acid, also indicates that it is possible to decouple kinetic effects from those on channel steady-state inactivation. If this holds true, it raises the possibility of modulating channel kinetics without generalized inhibitory effects, which would potentially lead to being able to fine-tune channel activity pharmacologically.
The rapid potentiation of N-type ICa by a subset of NAAN and arachidonic acid is an unusual effect, and may also indicate the existence of another largely uncharacterized site on these channels. Amphiphiles as a class do not tend to affect voltage-gated channel activation (Lundbaek, 2008), which indicates that the effects of the negatively charged NAAN and arachidonic acid are not simply non-specific. Arachidonic acid and NAAN (Barrett et al., 2001; Guo et al., 2008) likely affect N-type ICa activation at a site accessible from the outer leaflet of the plasma membrane, different to the site mediating increases in inactivation, which may be within the inner leaflet or be on an intracellular domain. Intriguingly, Δ9-THC has ‘arachidonic acid-like’ effects on CaV3.1 and CaV3.2; it potentiates currents at potentials where fewer channels are activated while producing a profound inhibition of maximally activated currents through an increase in steady-state inactivation (Ross et al., 2008). The observations that only some NAAN engage the putative potentiating site and that the potency to access the inhibitory site differs significantly among the small number of NAAN so far examined suggest that they may also be valuable tools to further define these two sites.
Actions of NAAN at potassium channels
Anandamide modulates a wide range of potassium channels including voltage dependent-, calcium activated- and two pore domain channels (reviewed in Oz, 2006). To date, only NA-Ser has been reported to mimic any of these effects (Godlewski et al., 2009), while NA-Gly and NA-GABA-OH have been shown not to affect human two pore domain TASK-1 channels (Barbara et al., 2009). In experiments on recombinant human BKCa (KCNMA1), NA-Ser produced a hyperpolarizing shift in the voltage-dependence of channel α-subunit activation (Godlewski et al., 2009), meaning outward current produced by a given depolarization was greater in the presence of NA-Ser. This effect was concentration-dependent (EC50∼ 3 µM), cannabinoid receptor-, G protein- and intracellular Ca2+ concentration-independent and mimicked by arachidonic acid and anandamide. Interestingly, the putative Abn-CBD receptor antagonist O-1918 had the opposite effect to NA-Ser; it shifted the activation curve for the BKCa channels strongly positive, blocking the effect of NA-Ser. These opposing and apparently receptor-independent effects of NA-Ser and O-1918 raise questions about the use of O-1918 to identify effects mediated by the putative Abn-CBD receptor, a possible site of action for anandamide, NA-DA and NA-Ser. For example, the NA-Ser mediated relaxation of intact rat mesenteric arterioles (Milman et al., 2006), which is O-1918-sensitive, superficially resembles effects of activating the Abn-CBD receptor, which had been previously suggested to mediate vessel relaxation by activating BKCa through Gi/Go proteins (Begg et al., 2003). However, the effects of NA-Ser on mesenteric arterioles are not pertussis toxin sensitive (Milman et al., 2006; Godlewski et al., 2009), and thus may be via direct NA-Ser modulation of BKCa.
Actions of NAANs at glycine transporters and receptors
NA-Gly is found in highest concentrations in the spinal cord (Huang et al., 2001), which is also where glycine acts as an inhibitory neurotransmitter. NA-Gly inhibits glycine transport by the glycine transporter GLYT2, but has no effect on the closely related glycine transporter GLYT1 or the GABA transporter GAT1. NA-Gly reduces the maximal rate of glycine transport but does not affect the Km for transport, which suggests that it is a non-competitive inhibitor (Wiles et al., 2006). NA-Gly inhibits transport with an IC50 of 5 µM, and NA-Ala has a similar potency and efficacy to NA-Gly. NA-GABA also inhibits GLYT2, albeit with significantly lower potency. It is interesting to note that although GLYT1 is insensitive to NA-Gly, transport activity is inhibited by arachidonic acid (IC50 ∼ 3 µM) and stimulated by high concentrations of anandamide (∼20 µM, Pearlman et al., 2003). By contrast, GLYT2 is insensitive to both arachidonic acid and anandamide. Thus, it appears that the chemical nature of the arachidonoyl conjugate head group plays an important role in determining the specificity of interactions with glycine transporters. The site of action of NA-Gly on GLYT2 has been investigated through the use of chimeric GLYT1/GLYT2 transporters and in contrast to the presumed membrane-delimited site of action of other NAANs, the recognition site appears to be on the extracellular surface of the protein (Edington et al., 2009). In particular, extracellular loops 2 and 4 of GLYT2 play crucial roles in forming the NA-Gly recognition site. A series of point mutations in extracellular loop 4 cause significant reductions in potency and efficacy of NA-Gly and NA-Ala. Extracellular loop 4 of GLYT2 is likely to play an important role in the gating process of the transporter by controlling access to the glycine binding site within the transporter (Ju et al., 2004, Singh et al., 2007) and thus, the mechanism of inhibition by NA-Gly may be to slow the gating process of the transporter (Edington et al., 2009).
NAANs as probes for the development of a novel pharmacology of glycine transporters
The differential sensitivity of glycine and GABA transporters to the activity of NA-Gly (and other closely related derivatives) provides an intriguing possibility that it may be possible to develop alternate compounds that mimic these effects to generate an alternative class of glycine transport inhibitors. Indeed, specific amino acid residues in extracellular loop 4 that are present in GLYT2 and not other closely related transporters have been identified that influence NA-Gly activity (Figure 2) (Edington et al., 2009). More detailed definition of how NA-Gly interacts with this site may provide design principles for developing novel synthetic compounds.
NA-Gly also modulates the activity of glycine receptors in a complex manner. At low concentrations of glycine, 10 µM NA-Gly stimulates the channel activity of glycine receptors, but at higher concentrations of glycine, the same concentration of NA-Gly speeds the rate of desensitization of the channel and reduces the steady-state current. Furthermore, the actions of NA-Gly are dependent on the time of exposure to the receptors (Yang et al., 2008).
Conclusions
NAAN interact with a diverse range of proteins, and often do so in a manner subtly distinct from the well-described effects of molecules such as arachidonic acid or anandamide. The wide range of possible ‘headgroup’ moieties of NAAN make them potentially valuable pharmacological tools to probe poorly described binding sites on channels, receptors and transporters. There are already some intriguing questions raised by NAAN modulation of calcium channels and glycine transporters. It will be of interest to see how this field develops and whether therapeutic drugs can be developed from our growing understanding of the diverse actions of this class of compounds.
Acknowledgments
MC was supported by Australian Research Council Discovery Grant DP0878255; CWV and RJV were supported by NHMRC Project Grant 402564.
Glossary
Abbreviations
- Abn-CBD
abnormal cannabidiol
- COX2
cyclooxygenase 2
- FAAH
fatty acid amide hydrolase
- GAT1
γ-amino butyric acid transporter 1
- GLYT1
glycine transporter 1
- GLYT2
glycine transporter 2
- GPCR
G protein coupled receptor
- ICa
voltage gated calcium channel current
- NAc-Tau
N-acyl taurine
- NAAN
N-acyl amino acid/neurotransmitter
- NA-Ala
N-arachidonoyl alanine
- NA-DA
N-arachidonoyl dopamine
- NA-GABA
N-arachidonoyl γ-amino butyric acid
- NA-Gly
N-arachidonoyl glycine
- NA-Ser
N-arachidonoyl serine
- NA-5HT
N-arachidonoyl serotonin
- NA-Tau
N-arachidonoyl taurine
- NO-DA
N-oleoyl dopamine
- NP-DA
N-palmitoyl dopamine
- NP-Gly
N-palmitoyl glycine
- NS-DA
N-stearoyl dopamine
- PGH2-Gly
prostaglandin H2-glycine
- TRP
transient receptor potential channel
- TRPM8
transient receptor potential melastatin 8 receptor
- TRPV1
transient receptor potential vanilloid 1 receptor
- TRPV4
transient receptor potential vanilloid 4 receptor
Conflict of interest
The authors declare that they have no conflicts of interest related to this work. Each author has made written contributions to this manuscript, and has approved the final version.
References
- Ahern GP. Activation of TRPV1 by the satiety factor oleoylethanolamide. J Biol Chem. 2003;278:30429–30434. doi: 10.1074/jbc.M305051200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S101. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasi R, Szoke E, Bolcskei K, Varga A, Riedl Z, Sandor Z, et al. Actions of 3-methyl-N-oleoyldopamine, 4-methyl-N-oleoyldopamine and N-oleoylethanolamide on the rat TRPV1 receptor in vitro and in vivo. Life Sci. 2008;82:644–671. doi: 10.1016/j.lfs.2007.12.022. [DOI] [PubMed] [Google Scholar]
- Aneetha H, O'Dell DK, Tan B, Walker JM, Hurley TD. Alcohol dehydrogenase-catalysed in vitro oxidation of anandamide to N-arachidonoyl glycine, a lipid mediator: synthesis of N-acyl glycinals. Bioorg Med Chem Lett. 2009;19:237–241. doi: 10.1016/j.bmcl.2008.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara G, Alloui A, Nargeot J, Lory P, Eschalier A, Bourinet E, et al. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J Neurosci. 2009;29:13106–13114. doi: 10.1523/JNEUROSCI.2919-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CF, Liu L, Rittenhouse AR. Arachidonic acid reversibly enhances N-type calcium current at an extracellular site. Am J Physiol Cell Physiol. 2001;280:1306–1318. doi: 10.1152/ajpcell.2001.280.5.C1306. [DOI] [PubMed] [Google Scholar]
- Begg M, Mo F-M, Offertaler L, Batkai S, Pacher P, Razdan RK, et al. G protein-coupled endothelial receptor for atypical cannabinoid ligands modulates a Ca2+-dependent K+ current. J Biol Chem. 2003;278:46188–46194. doi: 10.1074/jbc.M307258200. [DOI] [PubMed] [Google Scholar]
- Bezuglov V, Bobrov M, Gretskaya N, Gonchar A, Zinchenko G, Melck D, et al. Synthesis and biological evaluation of novel amides of polyunsaturated fatty acids with dopamine. Bioorg Med Chem Lett. 2001;11:447–449. doi: 10.1016/s0960-894x(00)00689-2. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Melck D, Bobrov MY, Gretskaya NM, Bezuglov VV, De Petrocellis L, et al. N-acyl-dopamines: novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem J. 2000;351:817–824. [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Krey JF, Walker JM. Sex and hormonal cycle differences in rat brain levels of pain-related cannabimimetic lipid mediators. Am J Physiol Regul Integr Comp Physiol. 2006;291:R349–R358. doi: 10.1152/ajpregu.00933.2005. [DOI] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS-J, Benton VM, Stuart JM, Masuda K, et al. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009a;10:14. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS-J, Burstein S, Walker JM. Novel endogenous N-acyl glycines: identification and characterization. In: Gerald Litwack G, editor. Vitamins Hormones, Volume 81: Anandamide An Endogenous Cannabinoid. London: Academic Press; 2009b. pp. 192–205. [DOI] [PubMed] [Google Scholar]
- Burstein SH, Rossetti RG, Yagen B, Zurier RB. Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat. 2000;61:29–41. doi: 10.1016/s0090-6980(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Burstein SH, Huang SM, Petros TJ, Rossetti RG, Walker JM, Zurier RB. Regulation of anandamide tissue levels by N-arachidonylglycine. Biochem Pharm. 2002;64:1147–1150. doi: 10.1016/s0006-2952(02)01301-1. [DOI] [PubMed] [Google Scholar]
- Burstein SH, Adams JK, Bradshaw HB, Fraioli C, Rossetti RG, Salmonsen RA, et al. Potential anti-inflammatory actions of the elmiric (lipoamino) acids. Bioorg Med Chem. 2007;15:3345–3355. doi: 10.1016/j.bmc.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Harris TJ, Kreek MJ. Topical capsaicin-induced allodynia in unanaesthetized primates: pharmacological modulation. J Pharmacol Exp Ther. 2003;306:1106–1114. doi: 10.1124/jpet.103.052381. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek MJ. Antiallodynic effects of loperamide and fentanyl against topical capsaicin-induced allodynia in unanaesthetized primates. J Pharmacol Exp Ther. 2004;311:155–163. doi: 10.1124/jpet.104.068411. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Minassi A, Ligresti A, Appendino G, Burstein S, Di Marzo V. A structure activity relationship on N-arachidonoyl amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Comm. 2004;314:192–196. doi: 10.1016/j.bbrc.2003.12.075. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001;20:7033–7040. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Nargeot J, Lory P. Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J Biol Chem. 2007;282:2314–2323. doi: 10.1074/jbc.M610033200. [DOI] [PubMed] [Google Scholar]
- Chu CJ, Huang SM, De Petrocellis L, Bisogno T, Ewing SA, Miller JD, et al. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J Biol Chem. 2003;278:13633–13639. doi: 10.1074/jbc.M211231200. [DOI] [PubMed] [Google Scholar]
- Chu Z-L, Carroll C, Chen R, Alfonso J, Gutierrez V, He H, et al. N-oleoyldopamine enhances glucose homeostasis through activation of GPR119. Mol Endocrinol. 2010;24:161–170. doi: 10.1210/me.2009-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Marzo D. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Chu CJ, Moriello AS, Kellner JC, Walker JM, Di Marzo V. Actions of two naturally occurring saturated N-acyldopamines on transient receptor potential vanilloid (TRPV1) channels. Br J Pharmacol. 2004;143:251–256. doi: 10.1038/sj.bjp.0705924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Starowicz K, Moriello AS, Vivese M, Orlando P, Di Marzo V. Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): effect of cAMP, cannabinoid CB1 receptors and endovanilloids. Exp Cell Res. 2007;313:1911–1920. doi: 10.1016/j.yexcr.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Edington AR, McKinzie AA, Reynolds AJ, Kassiou M, Ryan RM, Vandenberg RJ. Extracellular loops 2 and 4 of GLYT2 are required for N-arachidonylglycine inhibition of glycine transport. J Biol Chem. 2009;284:36424–36430. doi: 10.1074/jbc.M109.017509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ. Plant-derived, synthetic and endogenous cannabinoids as neuroprotective agents: non-psychoactive cannabinoids, ‘entourage’ compounds and inhibitors of N-acyl ethanolamine breakdown as therapeutic strategies to avoid psychotropic effects. Brain Res Rev. 2003;41:26–43. doi: 10.1016/s0165-0173(02)00218-7. [DOI] [PubMed] [Google Scholar]
- Godlewski G, Offertaler L, Osei-Hyiaman D, Mo FM, Harvey-White J, Liu J, et al. The endogenous brain constituent N-arachidonoyl L-serine is an activator of large conductance Ca2+-activated K+ channels. J Pharmacol Exp Ther. 2009;328:351–361. doi: 10.1124/jpet.108.144717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Ikeda SR. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol. 2004;65:665–674. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- Guo J, Williams DJ, Ikeda SR. N-arachidonyl serine, a putative endocannabinoid, alters the activation of N-type calcium channels in sympathetic neurons. J Neurophysiol. 2008;100:1147–1151. doi: 10.1152/jn.01204.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, et al. IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucl Acids Res. 2009;37:D680–D685. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herradon E, Martin MI, Lopez-Miranda V. Characterization of the vasorelaxant mechanisms of the endocannabinoid anandamide in rat aorta. Br J Pharmacol. 2007;152:699–708. doi: 10.1038/sj.bjp.0707404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SS-J, Bradshaw HB, Benton VM, Chen JS-C, Huang SM, Minassi A, et al. The biosynthesis of N-arachidonoyl dopamine (NADA), a putative endocannabinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot Essent Fatty Acids. 2009;81:291–301. doi: 10.1016/j.plefa.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Petros TJ, Chang S-Y, Zavitsanos PA, Zipkin RE, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem. 2001;276:42639–42634. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, de Petrocellis L, Fezza F, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid receptors. Proc Natl Acad Sci USA. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci USA. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA, et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol. 2007;152:825–831. doi: 10.1038/sj.bjp.0707419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt S-E, Julius D. Molecular basis for species-specific sensitivity to ‘hot’ chili peppers. Cell. 2002;108:421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- Ju P, Aubrey KR, Vandenberg RJ. Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J Biol Chem. 2004;279:22983–22991. doi: 10.1074/jbc.M312484200. [DOI] [PubMed] [Google Scholar]
- Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, et al. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Comm. 2006;347:827–832. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Liu L, Barrett CF, Rittenhouse AR. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am J Physiol Cell Physiol. 2001;280:1293–1305. doi: 10.1152/ajpcell.2001.280.5.C1293. [DOI] [PubMed] [Google Scholar]
- Lundbaek JA. Lipid bilayer-mediated regulation of ion channel function by amphiphilic drugs. J Gen Physiol. 2008;131:421–429. doi: 10.1085/jgp.200709948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue JM, Driscoll WJ, Mueller GP. Cytochrome c catalyzes the in vitro synthesis of arachidonoyl glycine. Biochem Biophys Res Comm. 2008;365:322–327. doi: 10.1016/j.bbrc.2007.10.175. [DOI] [PubMed] [Google Scholar]
- McHugh D, Tanner C, Mechoulam R, Pertwee RG, Ross RA. Inhibition of human neutrophil chemotaxis by endogenous cannabinoids and phytocannabinoids: evidence for a site distinct from CB1 and CB2. Mol Pharmacol. 2008;73:441–450. doi: 10.1124/mol.107.041863. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Florenzano F, Fezza F, Viscomi MT, van der Stelt M, et al. N-arachidonoyl-dopamine tunes synaptic transmission onto dopaminergic neurons by activating both cannabinoid and vanilloid receptors. Neuropsychopharmacology. 2007;32:298–308. doi: 10.1038/sj.npp.1301118. [DOI] [PubMed] [Google Scholar]
- Merkler DJ, Chew GH, Gee AJ, Merkler KA, Sorondo J-PO, Johnson ME. Oleic acid derived metabolites in mouse neuroblastoma N18TG2 cells. Biochemistry. 2004;43:12667–12674. doi: 10.1021/bi049529p. [DOI] [PubMed] [Google Scholar]
- Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Baktai S, et al. N-arachidonyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci USA. 2006;103:2428–2433. doi: 10.1073/pnas.0510676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Yoon JM, Moon MJ, Hwang J-I, Choe H, Lee JE, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054–21604. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD. Characterisation of the vasorelaxant properties of the novel endocannabinoid N-arachidonoyl-dopamine (NADA) Br J Pharmacol. 2004;141:803–812. doi: 10.1038/sj.bjp.0705643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD. Vascular effects of Δ9-terahydrocannabinol (THC), anandamide and N-arachidonoyl dopamine in the rat isolated aorta. Eur J Pharmacol. 2005;507:211–221. doi: 10.1016/j.ejphar.2004.11.056. [DOI] [PubMed] [Google Scholar]
- Oz M. Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharm Ther. 2006;111:114–144. doi: 10.1016/j.pharmthera.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Parmar N, Ho W-SV. N-arachidonoyl glycine, an endogenous lipid that acts as a vasorelaxant via nitric oxide and large conductance calcium-activated potassium channels. Br J Pharmacol. 2010;160:594–603. doi: 10.1111/j.1476-5381.2009.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman RJ, Aubrey KR, Vandenberg RJ. Arachidonic acid and anandamide have opposite modulatory actions on the GLYT1 subtype of glycine transporters. J Neurochem. 2003;84:592–601. doi: 10.1046/j.1471-4159.2003.01549.x. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Astarita G, Rapaka R. A neuroscientist's guide to lipidomics. Nat Rev Neurosci. 2007;8:743–754. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Qi Z-H, van Buren J, Riasinghani M. Enhancement of potency and efficacy of NADA by PKC-mediated phosphorylation of vanilloid receptor. J Neurophysiol. 2004;91:1442–1449. doi: 10.1152/jn.00745.2003. [DOI] [PubMed] [Google Scholar]
- Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Selective oxygenation of N-arachidonoyl glycine by cyclooxygenase-2. Biochem Biophys Res Commun. 2002;296:612–617. doi: 10.1016/s0006-291x(02)00915-4. [DOI] [PubMed] [Google Scholar]
- Rimmerman N, Bradshaw HB, Hughes HV, Chen JS-C HSS-J, McHugh D, et al. N-Palmitoyl glycine, a novel endogenous lipid that acts as a modulator of calcium influx and nitric oxide production in sensory neurons. Mol Pharmacol. 2008;74:213–224. doi: 10.1124/mol.108.045997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmerman N, Bradshaw HB, Basnet A, Tan B, Widlanski TS, Walker JM. Microsomal omega-hydroxylated metabolites of N-arachidonoyl dopamine are active at recombinant human TRPV1 receptors. Prostaglandins Other Lipid Mediat. 2009;88:10–17. doi: 10.1016/j.prostaglandins.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HR, Napier I, Connor M. Inhibition of recombinant human T-type calcium channels by Δ9-Tetrahydrocannabinol and cannabidiol. J Biol Chem. 2008;283:16124–16134. doi: 10.1074/jbc.M707104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HR, Gilmore AJ, Connor M. Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonyl dopamine. Br J Pharmacol. 2009;156:740–750. doi: 10.1111/j.1476-5381.2008.00072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA. The enigmatic pharmacology of GPR55. Trends Neurosci. 2009;30:156–163. doi: 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siudzdak G, Cravatt BF. Assignment of endogenous substrates to enzymes by global metabolic profiling. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry. 2006;45:9007–9015. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- Seuwen K, Ludwig M-G, Wolf RM. Receptors for protons or lipid messengers or both. J Recep Sig Trans Res. 2006;26:599–610. doi: 10.1080/10799890600932220. [DOI] [PubMed] [Google Scholar]
- Sharkey KA, Cristino L, Oland LD, van Sickle MD, Starowicz K, Pittman QJ, et al. Arvanil, anandamide and N-arachidonoyl dopamine (NADA) inhibit emesis through cannabinoid CB1 and vanilloid TRPV1 receptors in the ferret. Eur J Neurosci. 2007;25:2773–2782. doi: 10.1111/j.1460-9568.2007.05521.x. [DOI] [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the rat brain cannabinoid receptor. J Med Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Sipe JC, Arbour N, Gerber A, Beutler E. Reduced endocannabinoid immune modulation by a common cannabinoid 2 (CB2) receptor gene polymorphism: possible risk for autoimmune disorders. J Leukoc Biol. 2005;78:231–238. doi: 10.1189/jlb.0205111. [DOI] [PubMed] [Google Scholar]
- Succar R, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat inflammatory pain model. Mol Pain. 2007;3:24. doi: 10.1186/1744-8069-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton KG, Garrett EM, Rutter AR, Bonnert TP, Jarolimek W, Seabrook GR. Functional characterization of the S512Y mutant vanilloid human TRPV1 receptor. Br J Pharmacol. 2005;146:702–711. doi: 10.1038/sj.bjp.0706356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsanyi J, Sandor Z, Petho G, Varga A, Bolcskei K, Almasi R, et al. Direct evidence for activation and desensitization of the capsaicin receptor by N-oleoyl dopamine on TRPV1-transfected cell line, in gene deleted mice and in the rat. Neurosci Lett. 2004;361:155–158. doi: 10.1016/j.neulet.2003.12.025. [DOI] [PubMed] [Google Scholar]
- Tan B, O'Dell DK, Yu YW, Monn MF, Hughes HV, Burstein S, et al. Identification of endogenous acyl amino acids based on a targeted lipidomics approach. J Lipid Res. 2010;51:112–119. doi: 10.1194/jlr.M900198-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C-F, Iwakami S, Mikajiri A, Shibuya M, Hanaoka F, Ebizuka Y, et al. Inhibition of in vitro prostaglandin and leukotriene synthesis by cinnamoyl-β-phenethylamine and N-acyldopamine derivatives. Chem Pharm Bull. 1992;40:396–400. doi: 10.1248/cpb.40.396. [DOI] [PubMed] [Google Scholar]
- Vuong LAQ, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat neuropathic pain model. Neuropharmacology. 2008;54:189–193. doi: 10.1016/j.neuropharm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Walpole CSJ, Wrigglesworth R, Bevan S, Campbell EA, Dray A, James IF, et al. Analogs of capsaicin with agonist activity as novel analgesic agents; structure-activity studies. 1. The aromatic ‘A-region’. J Med Chem. 1993;36:2362–2372. doi: 10.1021/jm00068a014. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Wiles AL, Pearlman RJ, Rosvall M, Aubrey K, Vandenberg RJ. N-arachidonyl glycine inhibits the glycine transporter, GLYT2a. J Neurochem. 2006;99:781–786. doi: 10.1111/j.1471-4159.2006.04107.x. [DOI] [PubMed] [Google Scholar]
- Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, et al. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidic acid receptor indicates a role in human platelet activation. J Biol Chem. 2009;284:17304–17319. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Aubrey KR, Alroy I, Harvey RJ, Vandenberg RJ, Lynch JW. Subunit-specific modulation of glycine receptors by cannabinoids and N-arachidonyl-glycine. Biochem Pharmacol. 2008;76:1014–1023. doi: 10.1016/j.bcp.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Shelton F, Otero F, Nguyen DG, Caldwell JS, et al. Lipid G protein-coupled receptor ligand identification using β-arrestin PathHunter™ assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]