

Abstract
The CNS can exhibit features of inflammation in response to injury, infection or disease, whereby resident cells generate inflammatory mediators, including cytokines, prostaglandins, free radicals and complement, chemokines and adhesion molecules that recruit immune cells, and activate glia and microglia. Cerebral ischaemia triggers acute inflammation, which exacerbates primary brain damage. The regulation of inflammation after stroke is multifaceted and comprises vascular effects, distinct cellular responses, apoptosis and chemotaxis. There are many cell types that are affected including neurons, astrocytes, microglia and endothelial cells, all responding to the resultant neuroinflammation in different ways. Over the past 20 years, researchers examining brain tissue at various time intervals after stroke observed the presence of inflammatory cells, neutrophils and monocytes at the site of injury, as well as the activation of endogenous glia and microglia. This review examines the involvement of these cells in the progression of neural injury and proposes that the Toll-like receptors (TLRs) are likely to be an integral component in the communication between the CNS and the periphery. This receptor system is the archetypal pathogen sensing receptor system and its presence and signalling in the brain following neural injury suggests a more diverse role. We propose that the TLR system presents excellent pharmacological targets for the design of a new generation of therapeutic agents to modulate the inflammation that accompanies neural injury.
Keywords: toll-like receptors, stroke, neural inflammation, T-cells, macrophages
The immune system and the CNS have traditionally been thought of as two separate entities, and any interaction between the two has been seen as a sign of malfunctioning systems. However, over the past few years, evidence for a relationship between the two systems, particularly in neural injury, has been building (Zierath et al., 2010). During neural injury, there are multiple cell types and considerably more inflammatory mediators activated and released, regardless of the origin of the injury. Resident cells, including microglia and astrocytes, primarily control the immediate response to neural injury; secondary to this there is recruitment of non-resident cells including macrophages. During traumatic and ischaemic neural injury, the integrity of the blood–brain barrier (BBB) is disrupted, leading to increased and often unregulated communication between the CNS and the immune system. However, communication between the two systems also occurs during injury that does not necessarily damage the BBB (Kaur and Ling, 2008). The precise methods of communication between the two systems is yet to be elucidated, however, we are starting to gather clues as to how the messages from the damaged cells are relayed to the wider circulation, regardless of the integrity of the BBB. This review will examine the responses of the various cell types to neuro-inflammation that is a part of most CNS injuries, with a particular focus on ischaemic injury, and then attempt to integrate this in to a cohesive view of possible methods of communication between the cell types involved. Finally this information will be used to summarize the pharmacological challenges and opportunities presented.
Pathology of stroke
The most common form of ischaemic injury in the brain is stroke, caused by the loss of blood flow to a specific region of the brain. This is most often the result of an embolism in one of the arteries supplying the brain (Bejot et al., 2009). The immediate result is a decrease in perfusion of the area previously supplied by these arteries, causing metabolic stress on resident cells, in particular, the neurons, which do not store ATP and have a high energy demand. This causes immediate death in the area where perfusion is most significantly decreased (Nishino et al., 1994). Surrounding this area is a significant proportion of tissue that suffers a reduction in perfusion and also reacts to the adjacent death of severely affected tissue (Globus et al., 1990), creating a core of terminally damaged tissue and a surrounding penumbra of inflamed and slowly dying tissue.
Cell death mechanisms
It has been suggested that there is a combination of both apoptosis and necrosis as a result of stroke. With regard to neuroprotection the timing and the type of cell death will have consequences for potential neuroprotective therapies. Early investigation in to the type of cell death has focused on models of ischaemic death, including the in vivo model of stroke middle cerebral artery occlusion (MCAO) and in vitro simulation of ischaemia by oxygen and glucose deprivation (OGD). Much of the early work focused on the time immediately following the occlusion and found that during this time the primary means of death was necrosis (Smith et al., 1984). Further research has found evidence for apoptosis (Smith et al., 1984; Li et al., 1995; Hata et al., 1999). More recently, the idea that neuronal death is both apoptotic and necrotic has been supported by post-mortem studies in humans that have suffered stroke (Love et al., 2000; Sairanen et al., 2009). It is now accepted that the initial cell death seen in stroke is necrotic and following prolonged ischaemia and subsequent reperfusion, apoptosis becomes the primary mode of cell death. The apoptosis generated by stroke has been shown in animals and humans to continue for days (Kirino, 1982; Petito et al., 1987), suggesting that there is potential to interfere pharmacologically with this ordered process and provide neuro-protection.
Necrosis
As mentioned earlier, neurons have a high metabolic rate and are reliant on oxidative metabolism, therefore when the oxygen supply is significantly decreased, the cells quickly suffer metabolic stress. As the mitochondria begin to function incorrectly the delicate redox balance is disrupted, causing a pro-oxidant state. As the reactive oxygen species (ROS) are created in the proximity of the mitochondria, it is here that they elicit most of their damage (Choi et al., 2009). The ROS attack the lipid bilayer that surrounds the mitochondria and cause the release of cytochrome-c through loss of membrane integrity, leading to the activation of the apoptosome through caspases (Ferrer et al., 2003). ROS produced during ischaemic stress can also cause necrotic death by making holes in the cell membrane and causing the loss of membrane potential. This in turn opens the calcium channels causing a massive increase in intracellular calcium and the cell subsequently dies (Levraut et al., 2003).
Apoptosis
Both the intrinsic and extrinsic apoptotic pathways are used to cause cell death following ischaemia. The intrinsic pathway is activated when the neurons are unable to maintain ion balance causing a loss of membrane potential, this in turn leads to a calcium influx causing a disruption of the mitochondria. This disruption induces the opening of the mitochondrial membrane, releasing cytochrome c, allowing the apoptosome to be formed and causing the apoptosis of the cell through the actions of caspase 9 and 3. It has been shown that inhibiting this pathway can lead to a decrease in the size of infarct following stroke (Guan et al., 2006). The same study also showed that by inhibiting the extrinsic pathway, the infarct size was decreased, suggesting that in the pathology of stroke, both pathways are activated. The extrinsic pathway is activated by the death ligand, FasL binding to the death receptor, Fas. This in turn causes the dimerization, cleavage and activation of caspase 8, leading to apoptosis. Further studies have also shown that inhibition of this pathway leads to increased primary neuronal survival following OGD and that tumour necrosis factor-α (TNF-α, neutralizing antibodies decreased infarct size in vivo (Martin-Villalba et al., 2001).
Recently mechanisms that do not fall in to the either of the categories of apoptosis or necrosis have been identified as playing a role in the neural injury seen after stroke. This suggests that there is continuum of cell death mechanisms, of which apoptosis and necrosis are the extremes. One group has labelled one of these forms ‘necroptosis’, which is a description of Fas receptor signalling that can activate alternative pathways to the extrinsic pathways, leading to autophagy (Degterev et al., 2005). Further work has shown that this type of death, autophagy, is important in cerebral ischaemia (Adhami et al., 2006). More detailed analyses of cell death mechanisms are provided in the following reviews (Bredesen et al., 2006; Blomgren et al., 2007; Degterev and Yuan, 2008).
While there are many ways that ischaemia can cause necrosis, apoptosis and other forms of cellular death in the affected neuronal tissue, there has been much work done to understand how this affects the surrounding, surviving cells. The penumbra that surrounds the dying core is exposed to the cytoplasm of cells dying, via apoptotic, necrotic and other means, creating a potentially severe inflammatory environment.
Inflammation
Ischaemia-induced death of adjacent cells produces local inflammation, including the recruitment of leukocytes, macrophages and the production of inflammatory mediators. The immediate inflammatory response is primarily co-ordinated by the resident microglia (Giulian and Vaca, 1993). These cells have been shown to be activated within minutes of the infarct (Banati et al., 1993). Adjacent cellular damage is detected by receptors such as the Toll-like receptors (TLRs) whose ligands include the intracellular components hyaluronic acid (Termeer et al., 2002), mRNA (Kariko et al., 2004a) and various heat shock proteins (Ohashi et al., 2000; Asea et al., 2002). The activation of microglia leads to the release of important inflammatory cytokines and chemokines including TNF-α (Lambertsen et al., 2005), various interleukins (ILs; Minami et al., 1992; Block et al., 2000), and the up-regulation of the TNF-α receptor (Lambertsen et al., 2007), suggesting that the production of TNF-α can act in an autocrine manner. The chemokines, CCL2 and CCL3, are also released at this time from the activated microglia, leading to the attraction of macrophages and monocytes (Cowell et al., 2002; Schilling et al., 2009).
The activation of microglia is a process that has not been clearly shown to either help or hinder the recovery from stroke. Some studies have shown that through TNF-α, microglia can actively protect neurons (Lambertsen et al., 2009) and that mice lacking the p55 receptor for TNF-α have an increased stroke volume (Gary et al., 1998). However, others have shown that over-expression of TNF-α is detrimental to stroke outcome (Pettigrew et al., 2008). The most likely answer is that the timing and the degree of the expression of the inflammatory cytokines is the most important determinant of stroke outcome. This increases the importance of understanding the timing of the endogenous response and, consequently, creates a difficult situation for researchers wanting to carefully modulate the expression of a particular cytokine, as the duration and the magnitude of cytokine expression, artificially induced or applied, will critically affect the results.
Recruited cells in stroke
Although microglia are able to respond to adjacent cellular stress engendering an activated phenotype, during the recovery phase of stroke, cells from the periphery are recruited to the CNS (Garcia et al., 1994). The release of IL-1β, TNF-α, CCL2 and CCL3 by microglia attracts the immune cell to the area that has been damaged. A recent and comprehensive study has examined the time over which the immune cells are attracted to the infarct (Gelderblom et al., 2009).
The first cells to arrive are the neutrophils and macrophages with the primary role of removing cellular debris and releasing further cytokines to further attract more immune cells to the site. These cells arrive within minutes and 24 h respectively (Petry et al., 2007; Yilmaz and Granger, 2008). As part of their action, potentially damaging molecules, such as ROS and nitric oxide are released, despite the apparent dual action of these cells, the majority of research has shown that decreasing attraction of leukocytes to the injury site is beneficial, suggesting that the detrimental aspects outweigh the beneficial (Hartl et al., 1996). Inhibition of the chemoattractant chemokine, CXCL8 (IL-8) decreases damage following stroke (Villa et al., 2007) and this is supported by earlier work showing that inhibition of the proteasome blocked NFkB activation and decreased leukocyte infiltration via reduced IL release and resulted in a better stroke outcome (Phillips et al., 2000).
The cells next attracted to the site of injury are the myeloid dendritic cells and these have been detected at the site of injury 24 h post occlusion (Felger et al., 2010). These cells use interferon-γ (IFN-γ) to attract T-cells to the injury (Gottfried-Blackmore et al., 2009). The presence of T-lymphocytes has been shown up to 3 days after infarct (Jander et al., 1995) and to be damaging in the response to stroke (Yilmaz et al., 2006).However, there is still insufficient information regarding the activity of T-regulatory cells (Tregs) with respect to the benefit or detriment of controlling the T-cell response in stroke.
Neutrophils
The neutrophils are recruited earliest in the minutes following cerebral ischaemia, and have been shown to remain with in the brain up to 15 days post-injury (Weston et al., 2007). Although decreased neutrophil numbers in the brain has been correlated with a better outcome, the methods used to decrease infiltration also have anti-inflammatory effects (Allahtavakoli et al., 2009). In humans, a high peripheral neutrophil count is associated with greater stroke damage (Buck et al., 2008). However, once again, the high neutrophil counts maybe a result of a greater infarct, rather then higher numbers of neutrophils increasing the infarct size. It has also been shown that decreasing neutrophil degranulation can lead to a decrease in stroke damage, as the matrix metalloproteinase (MMP)-9 that is released contributes to the breakdown of the BBB (Strbian et al., 2007; Cuadrado et al., 2008). Excessive attraction of neutrophils to the damaged area has been thought to produce micro-clots in the smaller blood vessels, increasing the period of hypoxia and therefore subsequent damage (del Zoppo and Mabuchi, 2003). However, recently, mice deficient in granulocyte-colony stimulating factor (G-CSF), a chemo-attractant for neutrophils have been shown to have an increased stroke volume and this was rescued with the application of exogenous G-CSF (Strecker et al., 2010). As well as contributing directly to the response to cerebral ischaemia, the neutrophils also attract other cell types.
Monocyte/macrophages
Early work into the role of this cell type showed that over expression of the chemokine CCL2 [(monocyte chemoattractant protein-1 (MCP-1)] was detrimental in stroke (Chen et al., 2003), and that deficiency in this protein lead to an improved stroke outcome (Hughes et al., 2002). More recently further work has confirmed the presence of high levels of chemokines in both the periphery and the brain following stroke, but it has been suggested that detection of macrophages in the brain represents activated microglia and not the accumulation of monocytes from the periphery (Denker et al., 2007). This has been disputed by an exhaustive study published early in 2009 that shows large increases in infiltrating cells in the most common animal model of stroke, MCAO (Gelderblom et al., 2009). Although the presence of these cells is well accepted, their roles following stroke remains somewhat clouded. Macrophages in the brain have been shown to release the inflammatory mediators TNF-α and IL-1β (Clausen et al., 2008). Although the exact consequences of the presence of these mediators following stroke remains to be discovered, it is generally accepted that IL-1β is neurotoxic and IL-1β antagonists applied after stroke have been shown to be beneficial (Mulcahy et al., 2003). TNF-α on the other hand has been suggested to have a dual role, with TNF-α inhibition causing a smaller infarct (Dawson et al., 1996). In contrast to this, mice lacking the p55 TNF-α receptor were shown to have a greater brain injury (Gary et al., 1998). This study also showed that removal of the p75 receptor had no effect on infarct size, suggesting that antagonist specificity is required. To add further confusion to the TNF-α story, different mouse strains behave differently in regard to TNF-α production following stroke (Lambertsen et al., 2002).
T cells
The attraction of T cells to the site of injury has been suggested to be a double edged sword. Cytotoxic T cells are capable of increasing the infarct size, by attacking both damaged tissue and healthy neighbouring tissue. T helper (Th) cells also add to the inflammatory reaction, with Th-1 cells releasing the pro-inflammatory mediators IL-2, IL-12, TNF-α and IFN-γ (Yilmaz et al., 2006) and Th-2 cells producing the anti-inflammatory cytokines IL-10 and IL-13 (Pelidou et al., 1999). The Tregs further add to the T cell response by controlling the immune response to ischaemic damage (Liesz et al., 2009b). The Tregs are thought to produce their neuroprotection through IL-10, a cytokine that has been shown to both modulate neuronal viability thresholds (Grilli et al., 2000) and reduce injury in rats following focal stroke (Spera et al., 1998). Liesz and colleagues showed that depletion of Tregs leads to increased damage following MCAO and that this phenotype could be recovered with the intracerebroventricular administration of IL-10 (Liesz et al., 2009b). Production of IL-10 by many cell types, including monocytes, Th-2 cells and Tregs, is dependent on the TLR signalling pathway in particular TLR4, myeloid differentiation factor-88 (MyD88) and TRIF-dependent signalling (Boonstra et al., 2006; Samarasinghe et al., 2006). IL-10 is thought to mediate its neuroprotective effects through modulation of IFN-γ and TNF-α. Depletion of Tregs leads to an increase in both of these inflammatory mediators following stroke and IFN-γ antagonists have been shown to decrease damage when administered up to 3 days post-stroke (Yilmaz et al., 2006; Liesz et al., 2009b), significantly extending the previously short window of therapeutic opportunity and providing a potential for treatment in the days post-stroke. IL-10 from Tregs inhibits the production of TNF-α by down-regulating MyD88 (Dagvadorj et al., 2008) suggesting that the regulation of TLR pathways is important for stroke recovery. It is becoming clear that although the TLRs have a central role to play, it may not always be on either the neuroprotective or neurototoxic side of the equation. The response to stroke does not only involve multiple cell types, signalling systems and mediators, but each of these components can contribute to both damage and protection, depending upon the time and magnitude of action. The remaining cells that are involved in the response to stroke have far reaching effects that extend beyond the immediate CNS response and further into the immune system.
Breakdown of the BBB
In a closed head model of injury, such as MCAO, the BBB should initially remain intact, however, the release of proteases can decrease the integrity of the BBB and the metabolic stress can alter the permeability of the endothelial cells. It has been shown that, following MCAO, there is an increase in the expression of MMPs (Rosenberg et al., 1998). Inhibition of these enzymes can not only keep the BBB intact, it can reduce injury (Fujimoto et al., 2008; Maddahi et al., 2009), suggesting that the initial attraction and infiltration of peripheral cells is damaging. There has also been a suggestion that the CNS responds to peripheral infection, altering the way the brain functions through the expression of TNF-α and microglial activation (Riazi et al., 2008). This suggests that, without any interruption to the integrity of the BBB, there is crosstalk between the peripheral immune system and microglia, demonstrating the existence of at least one-way and possibly two-way communication between the CNS and periphery. One proposal is that the TLRs act to transmit messages from one side of the BBB to the other, regarding pathogens and cellular stress (Singh and Jiang, 2004). TLR4 in particular is expressed on the circumventricular organs and its adjuvant CD14 is up-regulated following systemic LPS infection (Laflamme and Rivest, 2001). During the pathology of stroke, regulation of the BBB function and access to the brain could be an important pharmacological target and the TLRs may prove to be a key component of this puzzle.
Immune system down-regulation
While much of the research on stroke has focused on the immediate inflammatory processes that occur during the ischaemia and reperfusion phase, clinically, one of the most important secondary factors affecting mortality is post-stroke infection (Roth et al., 2001; Vargas et al., 2006). The severity of the infection in most cases correlates with the severity of the stroke, but it is difficult to determine which is cause and which is effect. It is unclear whether a large infarct causes a subsequent large suppression of the immune system and more serious consequences of an infection, or if a serious infection will significantly inhibit neuronal recovery resulting in a more severe infarct. An initial increase in immune cell numbers in the first 24 h after stroke (Offner et al., 2006a) is followed by a significant decrease in immune function in the days following stroke (Offner et al., 2006b; Liesz et al., 2009a), which is matched with an increase in Tregs. The Tregs are thought to cause spleen atrophy and drastically decrease the number of circulating immune cells. However, once again there is uncertainty, as splenic atrophy could be caused by neural stimulation from the damaged brain and the apparent increase in Tregs could be a symptom of their ability to survive splenic atrophy, causing further dampening of the immune system. Considering however, that immune suppression may in itself be a contributing factor to the severity of stroke, perhaps an increase in Tregs in the days and weeks, rather than hours, following stroke should be considered a factor to be modulated to provide optimum protection from infection and minimization of infarct. Systemic infection following stroke can lead to an increased core temperature, increased acidosis and contribute to excitotoxicty, all leading to a larger infarct (Reith et al., 1996).
Clinically, it is quite difficult to determine if the stroke causes increases infection severity or if a severe infection increases stroke damage. It is becoming accepted that the ischaemia, reperfusion and subsequent neural damage causes, at first a heightened immune response and secondarily to that, immune suppression. When put in the context of the possibility of autoimmunity, this hypothesis appears to have a better fit. As a result of the stroke, immune cells have access to the usually off-limits CNS and in order to prevent the development of autoimmunity, the immune system is suppressed. It is rare to find a stroke patient who subsequently develops autoimmune disease, however, multiple clinical studies have shown the presence of brain-specific antibodies following stroke (Bornstein et al., 2001; Gromadzka et al., 2001; Dambinova et al., 2003). It has also been shown in experimental models of multiple sclerosis that astrocytes are responsible for stimulating T cells to become regulatory and limit autoimmunity (Trajkovic et al., 2004). However, this has not been extended to the paradigm of stroke. In experimental neural trauma, the autoimmune reaction can be modulated to produce a better outcome. Mice that were tolerant to myelin basic protein (MBP) prior to MCAO and had concurrent LPS treatment suffered a smaller infarct than those unexposed to MBP and autoimmunity was prevented (Gee et al., 2008). It was also shown that pre-treatment with LPS dampened the cellular immune response to stroke, but this may be more to do with preconditioning the TLRs in both the CNS and immune system, thus obscuring the precise reason for neuroprotection (Rosenzweig et al., 2004). Many studies have suggested that autoimmunity is detrimental in stroke, as mice with B and T cells removed suffer a smaller infarct (Hurn et al., 2007). However, this approach does not account for the different stages of inflammation, nor does it account for the contribution of different types of T cells. Both the CNS and peripheral response could be co-ordinated by TLR signalling, in the immediate response of the microglia and also the delayed response of the various immune cells. TLR ligands are present immediately following the infarct and are secreted into the periphery following BBB breakdown. TLRs are currently being targeted for various immune therapies, including autoimmune disease states (Gomariz et al., 2010) and the link between these receptors and the autoimmune consequences of stroke could provide potential therapies to minimize secondary damage following stroke. The current situation appears to be that autoimmunity can be developed after stroke and unless there is a subsequent infection, the consequences are minimal. As far as clinical treatments go, however, understanding the underlying mechanisms that control the immune reaction to the CNS will need to be known in greater detail in order facilitate pharmacological development in this area. One signalling system that appears to play a role in both the CNS and the peripheral response to stroke are the TLRs. Given the potential for these receptors to be involved as a bridge between the CNS and the periphery during ischaemic insult, a brief overview of TLR signalling will follow.
TLRs: the bridge between the CNS and periphery
TLRs are capable of signalling through NFkB to produce chemokines such as CCL2 (MCP-1) (Ueda et al., 1994), CCL3 (MIP-1α) (Grove and Plumb, 1993) and CCL4 (MIP-1β) and a murine orthologue of CXCL2 (MIP-2) (Widmer et al., 1993). After detecting damage to the CNS, TLRs on microglia are activated to produce a host of chemokines that are then thought to attract monocytes, leukocytes and neutrophils to the site of injury. These cells then pass through the endothelial cells into the brain parenchyma, where they contribute to the pathogenesis of stroke.
TLR expression in the brain
As a receptor system that is integral to the induction of the immune system it is not surprising to find TLRs highly expressed in tissue such as the lung and spleen (Nishimura and Naito, 2005). The brain, compared with the rest of the body, is relatively protected from stimuli that provoke an immune response, however, these receptors respond to both exogenous and endogenous stress. With a high glucose demand, the brain has a lower threshold for decreased perfusion or oxygen supply and consequently must have mechanisms in place that are capable of responding to stress such as ischaemia-reperfusion. While mRNA for all TLRs can be detected in the brain it is TLR 2 and 4 that are most highly expressed (Nishimura and Naito, 2005). There is further specificity when individual cell types with in the brain are considered. Although most studies have been done in mice and as yet have primarily reported on mRNA, rather than the more pertinent protein levels, a few studies have reported varying TLR expression in human neurons, astrocytes and microglia. While only TLR 3 mRNA has been detected in astrocytes in humans (Farina et al., 2005), murine astrocytes express TLR 2, 4, 5 and 9 mRNA (Bowman et al., 2003). The same study was also able to detect TLR 2 and 4 protein expression in these cells. Although these results are quite recent and yet to be supported by substantial literature with regard to protein studies, the primary role of the astrocyte is to provide structural and metabolic support to the neural tissue, the microglia on the other hand are the resident ‘macrophages’ of the brain and as expected express many TLRs. While mRNA for TLRs 1 through 10 has been detected in mouse microglia (Olson and Miller, 2004) all except TLR 10 have been detected in human microglia (Jack et al., 2005). Therefore the microglia should be able to respond quickly to both endogenous and exogenous threats. In murine neurons, mRNA for all TLRs has been detected (Mishra et al., 2006), human neurons, however, appear to differ substantially, as only mRNA for TLRs 3 (Lafon et al., 2006) and 8 (Ma et al., 2006) has been detected. Cell-type variation in the expression of TLRs leads to a variation in the inflammatory response dependent on cell type. It has recently been shown that TLR 2 is responsible for part of the damaging inflammatory response that follows cerebral ischaemia (Lehnardt et al., 2007). Through studies that develop tolerance with a preconditioning TLR agoinst, it has been shown that decreased TLR activity can lead to decreased damage (Kariko et al., 2004b; Marsh et al., 2009).
TLRs on invading cells
Some of the first TLR expression studies were carried out on immune cells. TLR expression on these cells is important not only for pathogen recognition but also activation, suggesting that TLR expression and function on the invading cells in stroke is not trivial.
Haematopoietic cells have been shown to express mRNA for all the TLRs. In particular, monocytes have been shown to express TLRs 1, 2, 5, 6 and 8 mRNA (Muzio et al., 2000; Hornung et al., 2002), macrophages TLRs 1, 4 and 9 (Medzhitov et al., 1997; Muzio et al., 2000; An et al., 2002) and T cells 2, 4 and 8 (Xu et al., 2005). The varied expression amongst these cells suggests a diverse role for the TLRs in haematopoietic cell function. While these receptors are obviously capable of responding to exogenous and endogenous stress, research has shown that the TLRs and their associated pathways are involved in the differentiation and maturation of haematopoietic cells.
In Drosophila, the TOLL pathway that gives its name to the TLRs has been shown to be involved in the regulation and differentiation of haematopoietic cells (Qiu et al., 1998). This has since been translated to show that the TLRs are involved in human haematopoiesis in response to inflammation (Ueda et al., 2005). The maturation of dendritic cells is reliant on the MyD88 dependent TLR pathway, as MyD88 deficient mice do not develop this cell type (Nagai et al., 2006). Ligation of TLRs on immune cells causes an increase in cell number and activity. Given that during cerebral ischaemic injury, TLR ligands are released from the injured CNS into the periphery, this receptor family has a significant role to play in all the aspects of neuronal ischaemic injury.
TLR signalling following inflammation
The control of early CNS inflammation is a careful balancing act, as both too much and too little inflammation will lead to decreased or delayed recovery. This suggests that the TLRs are well placed to initiate the inflammatory response, as their activation leads to the release of both pro and anti-inflammatory mediators.
Depending on which of the 11 TLRs activated by the signalling will either be MyD88 dependent, independent or some receptors have the ability to activate both types of pathway. Understanding the modes and consequences of TLR signalling could lead to novel therapies that regulate both the CNS and peripheral response to stroke. Figure 1 illustrates the pathways of TLR signalling.
Figure 1.
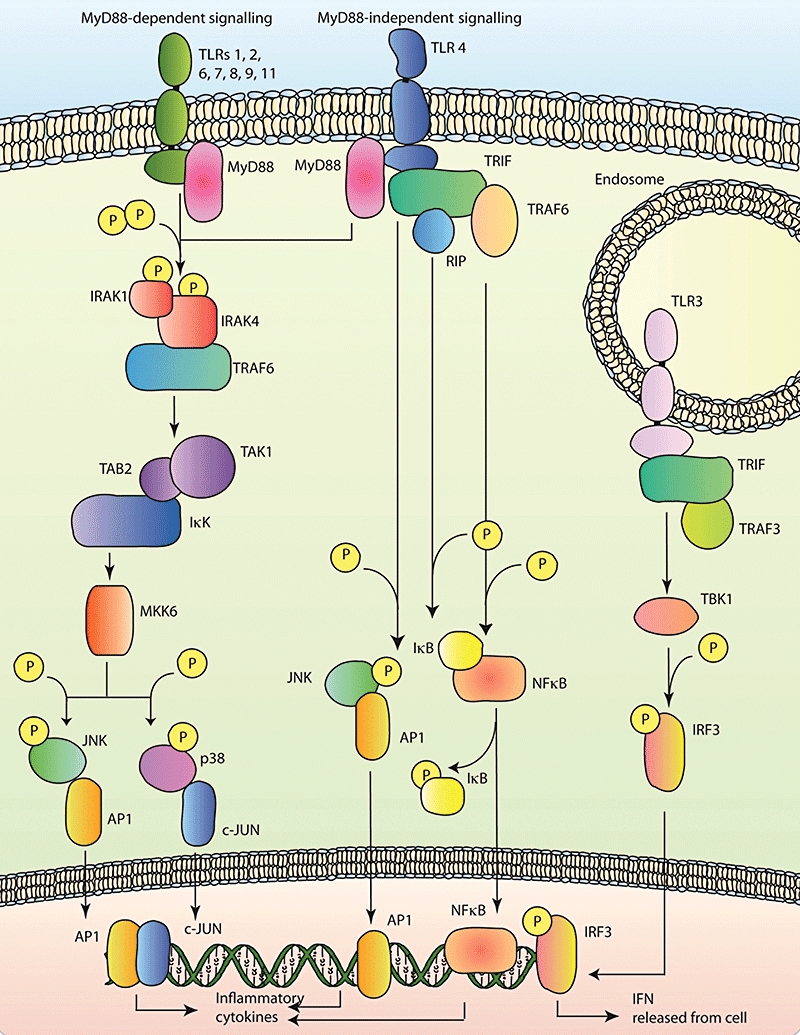
A diagram of the Toll-like receptor (TLR) pathways, indicating MyD88-dependent signalling on the left, MyD88-independent signalling on the right and TLR 3 only signalling on the upper right.
MyD88-dependent signalling
Upon ligand binding it is thought that a conformational change in the receptor allows association with adaptor proteins. Once MyD88 is bound to the receptor, IL receptor associated kinases (IRAK) 1 and 4 associate with MyD88 through the death domain, leading to phosporylation of both IRAKs (Wesche et al., 1997; Li et al., 2002). The IRAKs oligomerize with the tumour necrosis factor receptor associated factor-6 (TRAF-6), leading to activation and polyubiquitination of the oligomer itself (Deng et al., 2000). This oligomer then uses transforming growth factor β-activated kinase (TAK1), via the TAK1-binding protein (TAB2), to activate Ik B kinase (IKK). IKK is responsible for tagging IkB for degradation by phosphorylation and once IkB is degraded, NFkB is free to move to the nucleus and begin transcription of various inflammation-associated genes (Qian et al., 2001). TAK1, as well as activating IKK, is responsible for the activation of mitogen-activated kinase kinase 6 (MKK6) (Wang et al., 2001). MKK6 phosphorylates the kinases c-Jun N-terminal kinase (JNK) and p38, activation of these kinases leads to nuclear translocation and transcription of genes controlled by activator protein-1 (AP1) and c-JUN (Moriguchi et al., 1996). TLRs through MyD88 have been shown to have a role in many of the functions of the innate immune system, including leukocyte recruitment to CNS following traumatic injury (Babcock et al., 2008), wound healing (Macedo et al., 2007) and dendritic cell maturation and activation (Shen et al., 2008).
MyD88-independent signalling
TRIF was the third TLR adaptor discovered and pathways that are MyD88 independent rely on TRIF (Yamamoto et al., 2002). It was first thought that TRIF interacted with TRAF6 to induce NFkB and AP1 activation, however, TRAF6 knockout mice can still activate NFkB following activation of the MyD88 independent TLR3 (Meylan et al., 2004). TRIF binds to both TRAF6 and receptor interacting protein1 (RIP), which can both in turn activate NFkB pathways through interaction with IkB proteins. It was also shown that RIP was essential for TLR3 mediated JNK activation (Yamamoto et al., 2003). TRIF bound to TLR3 attracts TRAF3, causing the activation of TRAF family associated NFkB activator (TANK)-binding kinase or TBK1, this kinase then phosphorylates interferon regulatory factors (IRF), releasing interferons, thus attempting to combat the virus that activated TLR3 (Oganesyan et al., 2006). TRIF is the most important adaptor protein in the TLR response to viral infection, as TLR3 detects double stranded RNA from viruses and only uses this pathway.
The dual role for TLR signalling is exemplified by the discordance in TLR knockout studies of stroke. TLR2 has been shown to play an important role in the progression of neural injury following MCAO, a well-characterized model of stroke. This study showed that in the 96 h following stroke there was a 10-fold induction of TLR2 mRNA in the damaged hemisphere and that TLR2 knockout mice developed a smaller infarct (Lehnardt et al., 2007), this is supported by earlier research from Ziegler et al. (2007). However, in vitro experiments by another group have shown that microglial TLR4, and not TLR2, ligation is responsible for apoptosis (Jung et al., 2005). As these cells show a broad expression of TLRs, and multiple TLRs are activated when adjacent cells are dying, it is not surprising that studies in to TLR activation deliver contradictory findings. This result is typical of the complex role that these receptors and cells that express them play in the response to inflammation.
Further understanding of the role of the TLR signalling in the brain following stroke may lead to new pathways being identified to which potential therapeutics could be targeted. One potential target is the role that TLRs are now discovered to play in the phenomena of ischaemic preconditioning. An earlier and short exposure to ischaemia has been shown to produce a decreased infarct size without any pharmacological or genetic manipulations. The study of how the brain endogenously produces an improved outcome to stroke could guide pharmacological modulation. Three recent studies have shown that the TLRs are important modulators of the ischaemic tolerance phenotype (Hua et al., 2008; Stevens et al., 2008; Xing et al., 2008). The study targeting TLR 9 showed that TNF-α up-regulation was important for the neuronal protection afforded by preconditioning, as mice without TNF-α were unable to show a tolerant phenotype. These studies are beginning to map the cytokine environment that is required to produce the optimal amount of inflammation at the right time to produce the least damage following stroke. The understanding of this environment will be important in the design of new generation therapeutics.
Pharmacological challenges and opportunities
TLRs as specific drug targets
Given that the TLRs are involved in all aspects of the response to neuronal ischaemic injury, their potential for pharmacological modulation must be explored. Potential areas for intervention are summarized in Figure 2. These recently discovered receptors have already proven to be valuable in glioma treatment (Grauer et al., 2008) and as vaccine adjuvants (Cooper et al., 2004). The ability of TLR 8 to modulate the Treg response to injury and infection suggests that a synthetic agonist could enhance the Treg response while not activating other potentially damaging T cells (Peng et al., 2005). The induction of T helper cell type 1 is dependent on MyD88, while Th-2 cells are activated through MyD88 independent pathways (Lin et al., 2004), this presents an interesting pharmacological opportunity to direct Th cell type and create an infarct minimizing environment with the right TLR agonist. The potential for TLR based therapeutics in stroke is unlikely to be reached until there is further progress made in understanding the pharmacokinetics of TLR ligand binding and interactions between the TLR and the Toll/IL-1 receptor (TIR). The crystal structures of the ligand-binding domains for the TLR1 and 2 heterodimer, TLR3, TLR4 and TLR10 have been solved (Bell et al., 2005; Choe et al., 2005; Jin et al., 2007; Kim et al., 2007; Nyman et al., 2008). All of the structurally solved TLRs have also been shown to form dimers; TLR3 and 4 form homodimers and TLR1, heterodimers with TLRs 2 or 6, and only do so upon ligand binding (Gautam et al., 2006; Liu et al., 2008; Nyman et al., 2008; Park et al., 2009). This behaviour presents a further opportunity for pharmacological intervention by binding to the ligand-binding site or the dimerization site. Preventing dimerization may diminish signalling but not entirely inhibit it, and this ‘partial’ block maybe therapeutically better than complete block, as some TLR signalling has been shown to be beneficial while excessive activation can be detrimental. This approach may thus be able to strike a balance and deliver an infarct minimizing environment. The crystal structures of the TLRs have aided in successful drug design. However, in the response to stroke, although the TLRs are known to be involved, a better understanding of the underlying mechanisms needs to be developed before the potential for new therapies can be fulfilled.
Figure 2.
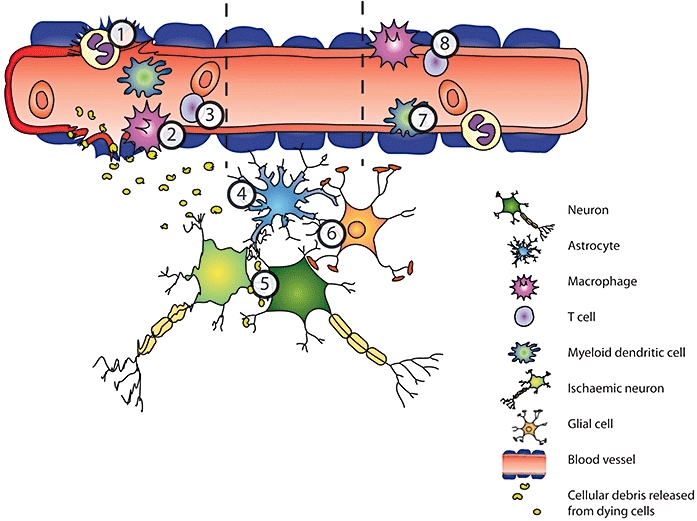
A diagrammatic representation of the cell types involved in the inflammatory response to stroke. The left hand side of the image shows cells migrating to the CNS from the periphery, where the integrity of the blood–brain barrier (BBB) is disrupted. The centre of the image shows the response of the resident cells and the right hand panel shows infiltrating cells regardless of BBB integrity. Each of the numbers refers to Toll-like receptor (TLR) involvement and potential for pharmacological intervention. 1.Neutrophils aTLR 4 deficiency has been shown to be important in the recruitment of neutrophils expressing myeloperoxidase (MPO), following injury (Kilic et al., 2008). 2.Macrophages aTLR expression increases in inflammatory conditions associated with cerebral vasculature, such as aneurysm, and the receptors have a role in activating macrophages in response to TNF-α (Jayaraman et al., 2008). 3.T -cells aTLR activation of microglia leads to activation of Th 1 cells (Jack et al., 2005). bTreg release of anti-inflammatory IL-10 is MyD88 and TRIF dependent (Boonstra et al., 2006). cRelease of IL-10 from Tregs leads to a down-regulation of MyD88 (Dagvadorj et al., 2008). dTLR 8 modulated the Treg response to injury (Peng et al., 2005). eTh1 cell induction is dependent on MyD88 pathways, while Th2 cells are activated via MyD88 independent pathways (Lin et al., 2004). 4.Astrocytes aTLR activation on astrocytes can lead to expression of MMP-9 and contribute to BBB breakdown (Hsieh et al., 2010). 5.Neurons aTLR 2 and 4 mediate neuronal death during stroke (Ziegler et al., 2007). 6.Glia aTLR 2 is expressed on microglia following ischemia and mice lacking TLR 2 suffer a smaller infarct (Lehnardt et al., 2007). bTLR 3 on glia releases neuroprotective mediators following inflammation (Bsibsi et al., 2006). 7.Myeloid dendritic cells aMyeloid related proteins 8 and 14 are thought to act as TLR ligands and increase recruitment of infiltrating myeloid cells (Ziegler et al., 2007). bIn response to inflammation TLR pathways mediate maturation (Ueda et al., 2005) of myeloid cells. 8.Endothelium aTLR 4 expression on the endothelium increases following sub-arachnoid haemorrhage (Zhou et al., 2007). bTLR ligands in the circulation lead to altered function of the BBB (Singh and Jiang, 2004). cTLR 4 on non-haematopoietic cells is important for controlling the CNS inflammatory response to peripheral stimuli (Chakravarty and Herkenham, 2005).
As yet there are few compounds that have shown potential in animal trials and subsequent efficacy in human trials in combating stroke. This is symptomatic of the complex nature of stroke pathology. Many of the processes and mechanisms involved are seemingly beneficial at one time or location and detrimental at another. Therefore it is unlikely that there will be a single solution that will be applicable to all patients at all treatment times, and to gain enough useful knowledge to be able to create the innovative solutions needed, we must use appropriate animal and cell culture models.
As demonstrated here the response to stroke is complex and not only involves multiple cell types but two systems in the body that are often considered the most separate. Translating the complexities of stroke to an in vitro set-up has inherent problems, but does not mean that in vitro studies are lacking in value. They provide an underpinning of knowledge concerning how individual parts of the puzzle act and how they relate to one another. Most importantly, these types of studies can be correlated with in vivo work to confirm the findings. In vivo work in this field is particularly important. Meta-analyses have revealed that experimental design may be the reason that so many treatments appear promising in animal models in vivo but fail to deliver in clinical trials (Macleod et al., 2004; Philip et al., 2009). Appropriate methodological guidelines are now available (Fisher et al., 2009) and should lead to more realistic expectations from research and preclinical trials. Another revelation from the meta-analysis was the time in which the treatments are given. Most stroke patients present some hours after the onset of symptoms, whereas in experimental systems, drug treatments are often given at the time of stroke or onset of reperfusion. When these candidate drugs are moved to clinical trials they are often given to patients in time windows significantly after they were shown to be effective in animals. The incredibly short window of opportunity for current treatments for stroke highlights this problem.
Secondary injury prevention
As ischaemic damage radiates from the core, there is a greater opportunity for pharmacological intervention to prevent secondary damage. Both the CNS and the immune system contribute to this damage, presenting two separate pharmaceutical targets. For the immune system, it has quite recently been reported that T cells infiltrate the brain and detrimentally produce cytokines; however, the infiltration can be decreased with the immunosuppressant FTY720 causing a reduction in infarct size (Shichita et al., 2009). Further development of this type of treatment will be interesting, as some T cells have been shown to beneficially enter the brain (Liesz et al., 2009b). It may become a case of inhibiting the detrimental, stimulating the beneficial or even both.
CNS-based treatments on the other hand have shown little promise with regard to diminishing delayed ischaemic damage. There are most likely a number of reasons for this; firstly there is inadequate fundamental understanding of the processes driven by the CNS that contribute to the extension of damage. It is only recently that TLRs have been revealed to play a significant role in not only the detection of neural damage but also the secondary, continuing cellular death. Second, the processes that are relatively well understood can be shown in various circumstances to be both beneficial and detrimental. The dual action of most pathways involved in this pathology causes significant problems for the timing of pharmacological intervention. Increasing the fundamental knowledge of the response to neuronal ischaemic injury will considerably increase the chances of overcoming both of these hurdles.
Further to the disappointing outcomes of many potential treatments has been the lack of effect in a clinical setting after promising animal and pre-clinical trials. Stroke is often related to cardiovascular disease, diabetes and old age (Fischer et al., 2006; Mulnier et al., 2006), whereas almost all experimental animals are young and healthy. When co-morbidities are included in the experimental design, it is rare that previously working treatments maintain their effectiveness (O'Collins et al., 2009). The reasons for this are most likely the increase in oxidative stress involved in the co-morbidities that therapies cannot overcome to engender neuroprotection, combined with an inability of the already damaged body to respond using the normal physiological response that some therapies attempt to alter.
Conclusion
Further research into the complex pathology of stroke will be helped with ideas derived from both neuroscience and immunology. It is the innate immunity that initializes the response to inflammation and cell death and primes the wider immune system to respond. Understanding the interplay between the CNS and the immune system will provide greater insight into the many pathologies that involve CNS inflammation and increase the number of potential pharmacological targets. In order to achieve this, innovative research methods will need to address the temporal specificity of activation of inflammatory mediators. The TLRs provide a tantalizing target, involved in many processes and cell types responding to stroke.
Acknowledgments
P.J.C. is funded by the NHMRC of Australia and by the National Heart Foundation of Australia.
Glossary
Abbreviations
- AP-1
activator protein-1
- BBB
blood–brain barrier
- G-CSF
granulocyte-colony stimulating factor
- IFN
interferon
- IKK
Ik B kinase
- IL
interleukin
- IRAK
interleukin receptor associated kinases
- IRF
interferon regulatory factor
- JNK
c-Jun N-terminal kinase
- MBP
myelin basic protein
- MCP-1
monocyte chemoattractant protein-1
- MCAO
middle cerebral artery occlusion
- MKK6
mitogen-activated kinase kinase 6
- MMP
matrix metalloproteinase
- MyD88
myeloid differentiation factor-88
- OGD
oxygen glucose deprivation
- RIP
receptor interacting protein
- ROS
reactive oxygen species
- TAB2
TAK1-binding protein
- TAK1
transforming growth factor β-activated kinase
- TANK
TRAF family associated NFkB activator
- TBK1
TANK-binding kinase
- TIR
Toll/interleukin-1 receptor
- TLR
Toll-like receptors
- TNF-α
tumour necrosis factor-α
- TRAF-6
tumour necrosis factor receptor associated factor-6
- Tregs
T-regulatory cells
- TRIF
TIR-domain-containing adapter-inducing interferon-β
Conflict of interest
The authors declare no conflict of interest.
References
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahtavakoli M, Moloudi R, Arababadi MK, Shamsizadeh A, Javanmardi K. Delayed post ischemic treatment with Rosiglitazone attenuates infarct volume, neurological deficits and neutrophilia after embolic stroke in rat. Brain Res. 2009;1271:121–127. doi: 10.1016/j.brainres.2009.03.040. [DOI] [PubMed] [Google Scholar]
- An H, Xu H, Yu Y, Zhang M, Qi R, Yan X, et al. Up-regulation of TLR9 gene expression by LPS in mouse macrophages via activation of NF-kappaB, ERK and p38 MAPK signal pathways. Immunol Lett. 2002;81:165–169. doi: 10.1016/s0165-2478(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Toft-Hansen H, Owens T. Signaling through MyD88 regulates leukocyte recruitment after brain injury. J Immunol. 2008;181:6481–6490. doi: 10.4049/jimmunol.181.9.6481. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- Bejot Y, Osseby GV, Gremeaux V, Durier J, Rouaud O, Moreau T, et al. Changes in risk factors and preventive treatments by stroke subtypes over 20 years: a population-based study. J Neurol Sci. 2009;287:84–88. doi: 10.1016/j.jns.2009.08.062. [DOI] [PubMed] [Google Scholar]
- Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, et al. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10976–10980. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block F, Peters M, Nolden-Koch M. Expression of IL-6 in the ischemic penumbra. Neuroreport. 2000;11:963–967. doi: 10.1097/00001756-200004070-00013. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007;12:993–1010. doi: 10.1007/s10495-007-0754-4. [DOI] [PubMed] [Google Scholar]
- Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, et al. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- Bornstein NM, Aronovich B, Korczyn AD, Shavit S, Michaelson DM, Chapman J. Antibodies to brain antigens following stroke. Neurology. 2001;56:529–530. doi: 10.1212/wnl.56.4.529. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck BH, Liebeskind DS, Saver JL, Bang OY, Yun SW, Starkman S, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39:355–360. doi: 10.1161/STROKEAHA.107.490128. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systematic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003;23:748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309:581–585. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- Choi K, Kim J, Kim GW, Choi C. Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. Curr Neurovasc Res. 2009;6:213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CL, Davis HL, Morris ML, Efler SM, Adhami MA, Krieg AM, et al. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J Clin Immunol. 2004;24:693–701. doi: 10.1007/s10875-004-6244-3. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- Cuadrado E, Ortega L, Hernandez-Guillamon M, Penalba A, Fernandez-Cadenas I, Rosell A, et al. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008;84:207–214. doi: 10.1189/jlb.0907606. [DOI] [PubMed] [Google Scholar]
- Dagvadorj J, Naiki Y, Tumurkhuu G, Hassan F, Islam S, Koide N, et al. Interleukin-10 inhibits tumor necrosis factor-alpha production in lipopolysaccharide-stimulated RAW 264.7 cells through reduced MyD88 expression. Innate Immun. 2008;14:109–115. doi: 10.1177/1753425908089618. [DOI] [PubMed] [Google Scholar]
- Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA. Blood test detecting autoantibodies to N-methyl-D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem. 2003;49:1752–1762. doi: 10.1373/49.10.1752. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci Lett. 1996;218:41–44. doi: 10.1016/0304-3940(96)13116-5. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Denker SP, Ji S, Dingman A, Lee SY, Derugin N, Wendland MF, et al. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J Neurochem. 2007;100:893–904. doi: 10.1111/j.1471-4159.2006.04162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Felger JC, Abe T, Kaunzner UW, Gottfried-Blackmore A, Gal-Toth J, McEwen BS, et al. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun. 2010;24:724–737. doi: 10.1016/j.bbi.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Friguls B, Dalfo E, Justicia C, Planas AM. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol Appl Neurobiol. 2003;29:472–481. doi: 10.1046/j.1365-2990.2003.00485.x. [DOI] [PubMed] [Google Scholar]
- Fischer U, Arnold M, Nedeltchev K, Schoenenberger RA, Kappeler L, Hollinger P, et al. Impact of comorbidity on ischemic stroke outcome. Acta Neurol Scand. 2006;113:108–113. doi: 10.1111/j.1600-0404.2005.00551.x. [DOI] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Takagi Y, Aoki T, Hayase M, Marumo T, Gomi M, et al. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1674–1685. doi: 10.1038/jcbfm.2008.59. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat) Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- Gautam JK, Ashish, Comeau LD, Krueger JK, Smith MF., Jr Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J Biol Chem. 2006;281:30132–30142. doi: 10.1074/jbc.M602057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008;39:1575–1582. doi: 10.1161/STROKEAHA.107.501486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Giulian D, Vaca K. Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke. 1993;24(12 Suppl):I84–I90. [PubMed] [Google Scholar]
- Globus MY, Busto R, Martinez E, Valdes I, Dietrich WD. Ischemia induces release of glutamate in regions spared from histopathologic damage in the rat. Stroke. 1990;21(11 Suppl):III43–III46. [PubMed] [Google Scholar]
- Gomariz RP, Gutierrez-Canas I, Arranz A, Carrion M, Juarranz Y, Leceta J, et al. Peptides targeting toll-like receptor signalling pathways for novel immune therapeutics. Curr Pharm Des. 2010;16:1063–1080. doi: 10.2174/138161210790963841. [DOI] [PubMed] [Google Scholar]
- Gottfried-Blackmore A, Kaunzner UW, Idoyaga J, Felger JC, McEwen BS, Bulloch K. Acute in vivo exposure to interferon-{gamma} enables resident brain dendritic cells to become effective antigen presenting cells. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0911509106. DOI: 10.1073/pnas.0911509106 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauer OM, Molling JW, Bennink E, Toonen LW, Sutmuller RP, Nierkens S, et al. TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol. 2008;181:6720–6729. doi: 10.4049/jimmunol.181.10.6720. [DOI] [PubMed] [Google Scholar]
- Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci. 2000;12:2265–2272. doi: 10.1046/j.1460-9568.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- Gromadzka G, Zielinska J, Ryglewicz D, Fiszer U, Czlonkowska A. Elevated levels of anti-heat shock protein antibodies in patients with cerebral ischemia. Cerebrovasc Dis. 2001;12:235–239. doi: 10.1159/000047709. [DOI] [PubMed] [Google Scholar]
- Grove M, Plumb M. C/EBP, NF-kappa B, and c-Ets family members and transcriptional regulation of the cell-specific and inducible macrophage inflammatory protein 1 alpha immediate-early gene. Mol Cell Biol. 1993;13:5276–5289. doi: 10.1128/mcb.13.9.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Liu XM, Wang XT, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006;1092:36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- Hartl R, Schurer L, Schmid-Schonbein GW, del Zoppo GJ. Experimental antileukocyte interventions in cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1108–1119. doi: 10.1097/00004647-199611000-00004. [DOI] [PubMed] [Google Scholar]
- Hata R, Gillardon F, Michaelidis TM, Hossmann KA. Targeted disruption of the bcl-2 gene in mice exacerbates focal ischemic brain injury. Metab Brain Dis. 1999;14:117–124. doi: 10.1023/a:1020709814456. [DOI] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002;22:308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15:42–51. doi: 10.1038/jcbfm.1995.5. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Paget A, Shin YS, Li X, Mayer J, Chaudhry H, et al. TNF-alpha-mediated inflammation in cerebral aneurysms: a potential link to growth and rupture. Vasc Health Risk Manag. 2008;4:805–817. doi: 10.2147/vhrm.s2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, et al. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004a;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling – a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004b;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res. 2008;5:71–81. doi: 10.2174/156720208783565645. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Lafon M, Megret F, Lafage M, Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J Mol Neurosci. 2006;29:185–194. doi: 10.1385/JMN:29:3:185. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Gregersen R, Finsen B. Microglial-macrophage synthesis of tumor necrosis factor after focal cerebral ischemia in mice is strain dependent. J Cereb Blood Flow Metab. 2002;22:785–797. doi: 10.1097/00004647-200207000-00004. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Fenger C, Wulf H, Owens T, Dagnaes-Hansen F, et al. Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience. 2007;144:934–949. doi: 10.1016/j.neuroscience.2006.10.046. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Meldgaard M, Ladeby R, Finsen B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:119–135. doi: 10.1038/sj.jcbfm.9600014. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–H558. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke. 1995;26:1252–1257. doi: 10.1161/01.str.26.7.1252. discussion 1257–1258. [DOI] [PubMed] [Google Scholar]
- Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009a;40:2849–2858. doi: 10.1161/STROKEAHA.109.549618. [DOI] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009b;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Lin L, Gerth AJ, Peng SL. CpG DNA redirects class-switching towards ‘Th1-like’ Ig isotype production via TLR9 and MyD88. Eur J Immunol. 2004;34:1483–1487. doi: 10.1002/eji.200324736. [DOI] [PubMed] [Google Scholar]
- Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Neuronal death in brain infarcts in man. Neuropathol Appl Neurobiol. 2000;26:55–66. doi: 10.1046/j.1365-2990.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo L, Pinhal-Enfield G, Alshits V, Elson G, Cronstein BN, Leibovich SJ. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol. 2007;171:1774–1788. doi: 10.2353/ajpath.2007.061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod MR, O'Collins T, Howells DW, Donnan GA. Pooling of animal experimental data reveals influence of study design and publication bias. Stroke. 2004;35:1203–1208. doi: 10.1161/01.STR.0000125719.25853.20. [DOI] [PubMed] [Google Scholar]
- Maddahi A, Chen Q, Edvinsson L. Enhanced cerebrovascular expression of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 via the MEK/ERK pathway during cerebral ischemia in the rat. BMC Neurosci. 2009;10:56. doi: 10.1186/1471-2202-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–1020. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Yabuuchi K, Yamazaki A, Satoh M. Induction of interleukin-1 beta mRNA in rat brain after transient forebrain ischemia. J Neurochem. 1992;58:390–392. doi: 10.1111/j.1471-4159.1992.tb09324.x. [DOI] [PubMed] [Google Scholar]
- Mishra BB, Mishra PK, Teale JM. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Kuroyanagi N, Yamaguchi K, Gotoh Y, Irie K, Kano T, et al. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol. 2003;140:471–476. doi: 10.1038/sj.bjp.0705462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulnier HE, Seaman HE, Raleigh VS, Soedamah-Muthu SS, Colhoun HM, Lawrenson RA, et al. Risk of stroke in people with type 2 diabetes in the UK: a study using the General Practice Research Database. Diabetologia. 2006;49:2859–2865. doi: 10.1007/s00125-006-0493-z. [DOI] [PubMed] [Google Scholar]
- Muzio M, Bosisio D, Polentarutti N, D'Amico G, Stoppacciaro A, Mancinelli R, et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000;164:5998–6004. doi: 10.4049/jimmunol.164.11.5998. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–892. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
- Nishino H, Czurko A, Fukuda A, Hashitani T, Hida H, Karadi Z, et al. Pathophysiological process after transient ischemia of the middle cerebral artery in the rat. Brain Res Bull. 1994;35:51–56. doi: 10.1016/0361-9230(94)90215-1. [DOI] [PubMed] [Google Scholar]
- Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund P. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283:11861–11865. doi: 10.1074/jbc.C800001200. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Donnan GA, Macleod MR, Howells DW. Scope of preclinical testing versus quality control within experiments. Stroke. 2009;40:e497. doi: 10.1161/STROKEAHA.109.550335. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006a;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006b;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Pelidou SH, Kostulas N, Matusevicius D, Kivisakk P, Kostulas V, Link H. High levels of IL-10 secreting cells are present in blood in cerebrovascular diseases. Eur J Neurol. 1999;6:437–442. doi: 10.1046/j.1468-1331.1999.640437.x. [DOI] [PubMed] [Google Scholar]
- Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- Petry KG, Boiziau C, Dousset V, Brochet B. Magnetic resonance imaging of human brain macrophage infiltration. Neurotherapeutics. 2007;4:434–442. doi: 10.1016/j.nurt.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew LC, Kindy MS, Scheff S, Springer JE, Kryscio RJ, Li Y, et al. Focal cerebral ischemia in the TNFalpha-transgenic rat. J Neuroinflammation. 2008;5:47. doi: 10.1186/1742-2094-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Benatar M, Fisher M, Savitz SI. Methodological quality of animal studies of neuroprotective agents currently in phase II/III acute ischemic stroke trials. Stroke. 2009;40:577–581. doi: 10.1161/STROKEAHA.108.524330. [DOI] [PubMed] [Google Scholar]
- Phillips JB, Williams AJ, Adams J, Elliott PJ, Tortella FC. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke. 2000;31:1686–1693. doi: 10.1161/01.str.31.7.1686. [DOI] [PubMed] [Google Scholar]
- Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFkappa B. J Biol Chem. 2001;276:41661–41667. doi: 10.1074/jbc.M102262200. [DOI] [PubMed] [Google Scholar]
- Qiu P, Pan PC, Govind S. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development. 1998;125:1909–1920. doi: 10.1242/dev.125.10.1909. [DOI] [PubMed] [Google Scholar]
- Reith J, Jorgensen HS, Pedersen PM, Nakayama H, Raaschou HO, Jeppesen LL, et al. Body temperature in acute stroke: relation to stroke severity, infarct size, mortality, and outcome. Lancet. 1996;347:422–425. doi: 10.1016/s0140-6736(96)90008-2. [DOI] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA. 2008;105:17151–17156. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Roth EJ, Lovell L, Harvey RL, Heinemann AW, Semik P, Diaz S. Incidence of and risk factors for medical complications during stroke rehabilitation. Stroke. 2001;32:523–529. doi: 10.1161/01.str.32.2.523. [DOI] [PubMed] [Google Scholar]
- Sairanen T, Szepesi R, Karjalainen-Lindsberg ML, Saksi J, Paetau A, Lindsberg PJ. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009;118:541–552. doi: 10.1007/s00401-009-0559-3. [DOI] [PubMed] [Google Scholar]
- Samarasinghe R, Tailor P, Tamura T, Kaisho T, Akira S, Ozato K. Induction of an anti-inflammatory cytokine, IL-10, in dendritic cells after toll-like receptor signaling. J Interferon Cytokine Res. 2006;26:893–900. doi: 10.1089/jir.2006.26.893. [DOI] [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161:806–812. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol. 2008;181:1849–1858. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology. 2004;201:197–207. doi: 10.1016/j.tox.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Smith ML, Auer RN, Siesjo BK. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol. 1984;64:319–332. doi: 10.1007/BF00690397. [DOI] [PubMed] [Google Scholar]
- Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett. 1998;251:189–192. doi: 10.1016/s0304-3940(98)00537-0. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strbian D, Karjalainen-Lindsberg ML, Kovanen PT, Tatlisumak T, Lindsberg PJ. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation. 2007;116:411–418. doi: 10.1161/CIRCULATIONAHA.106.655423. [DOI] [PubMed] [Google Scholar]
- Strecker JK, Sevimli S, Schilling M, Klocke R, Nikol S, Schneider A, et al. Effects of G-CSF treatment on neutrophil mobilization and neurological outcome after transient focal ischemia. Exp Neurol. 2010;222:108–113. doi: 10.1016/j.expneurol.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic V, Vuckovic O, Stosic-Grujicic S, Miljkovic D, Popadic D, Markovic M, et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia. 2004;47:168–179. doi: 10.1002/glia.20046. [DOI] [PubMed] [Google Scholar]
- Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153:2052–2063. [PubMed] [Google Scholar]
- Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas M, Horcajada JP, Obach V, Revilla M, Cervera A, Torres F, et al. Clinical consequences of infection in patients with acute stroke: is it prime time for further antibiotic trials? Stroke. 2006;37:461–465. doi: 10.1161/01.STR.0000199138.73365.b3. [DOI] [PubMed] [Google Scholar]
- Villa P, Triulzi S, Cavalieri B, Di Bitondo R, Bertini R, Barbera S, et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol Med. 2007;13:125–133. doi: 10.2119/2007-00008.Villa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CD, Chang GD, Lee YK, Chen H. A functional composite cis-element for NF kappa b and RBJ kappa in the rat pregnancy-specific glycoprotein gene. Biol Reprod. 2001;65:1437–1443. doi: 10.1095/biolreprod65.5.1437. [DOI] [PubMed] [Google Scholar]
- Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27:100–114. doi: 10.1038/sj.jcbfm.9600324. [DOI] [PubMed] [Google Scholar]
- Widmer U, Manogue KR, Cerami A, Sherry B. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J Immunol. 1993;150:4996–5012. [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, et al. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke. 2008;39:2362–2369. doi: 10.1161/STROKEAHA.107.507939. [DOI] [PubMed] [Google Scholar]
- Xu D, Komai-Koma M, Liew FY. Expression and function of Toll-like receptor on T cells. Cell Immunol. 2005;233:85–89. doi: 10.1016/j.cellimm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30:783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zhou ML, Shi JX, Hang CH, Zhang FF, Gao J, Yin HX. Expression of toll-like receptor 4 in the brain in a rabbit experimental subarachnoid haemorrhage model. Inflamm Res. 2007;56:93–97. doi: 10.1007/s00011-006-6035-9. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- Zierath D, Thullbery M, Hadwin J, Gee JM, Savos A, Kalil A, et al. CNS immune responses following experimental stroke. Neurocrit Care. 2010;12:274–284. doi: 10.1007/s12028-009-9270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]