

Abstract
Background and purpose:
Carboxylesterases (CEs) metabolize a wide range of xenobiotic substrates including heroin, cocaine, meperidine and the anticancer agent CPT-11. In this study, we have purified to homogeneity human liver and intestinal CEs and compared their ability with hydrolyse heroin, cocaine and CPT-11.
Experimental approach:
The hydrolysis of heroin and cocaine by recombinant human CEs was evaluated and the kinetic parameters determined. In addition, microsomal samples prepared from these tissues were subjected to chromatographic separation, and substrate hydrolysis and amounts of different CEs were determined.
Key results:
In contrast to previous reports, cocaine was not hydrolysed by the human liver CE, hCE1 (CES1), either as highly active recombinant protein or as CEs isolated from human liver or intestinal extracts. These results correlated well with computer-assisted molecular modelling studies that suggested that hydrolysis of cocaine by hCE1 (CES1), would be unlikely to occur. However, cocaine, heroin and CPT-11 were all substrates for the intestinal CE, hiCE (CES2), as determined using both the recombinant protein and the tissue fractions. Again, these data were in agreement with the modelling results.
Conclusions and implications:
These results indicate that the human liver CE is unlikely to play a role in the metabolism of cocaine and that hydrolysis of this substrate by this class of enzymes is via the human intestinal protein hiCE (CES2). In addition, because no enzyme inhibition is observed at high cocaine concentrations, potentially this route of hydrolysis is important in individuals who overdose on this agent.
Keywords: carboxylesterase, cocaine, heroin, CPT-11, inhibitor, molecular modelling
Introduction
Carboxylesterases [carboxylic ester hydrolase (CE), EC 3.1.1.1] are responsible for the detoxification of numerous xenobiotics including heroin, cocaine, meperidine and lidocaine, as well as the activation of the anticancer agents, capecitabine and CPT-11 (Pindel et al., 1997; Danks et al., 1999; Zhang et al., 1999; Tabata et al., 2004). CEs tend to be expressed in tissues likely to be exposed to such agents, for example, the liver, gut, kidney, lung. Although numerous CEs have been identified in rodents and other mammals, currently only three such enzymes have been characterized from humans. Human liver CE (hCE1; CES1) is predominantly expressed in the liver and is efficient at metabolizing small esterified substrates (Wadkins et al., 2001). Human intestinal CE (hiCE, hCE2, CES2) is expressed in the epithelial lining of the gut and the liver, and has been demonstrated to be efficient in the hydrolysis of the anticancer prodrug CPT-11 (Humerickhouse et al., 2000; Khanna et al., 2000). A third human CE, human brain CE (hBr3; CES3) has been recently identified, but as yet little is known about its pattern of expression and/or substrate specificity (Mori et al., 1999).
Cocaine and heroin abuse is a major clinical problem in the 25 to 49 year-old age group, with drug overdose being the fourth leading cause of death, on a par with motor vehicle accidents (Anderson, 1999). Hence devising alternative treatment options for patients who have overdosed on these agents is desirable. Although naloxone has significantly reduced the incidence of heroin-induced fatalities, there are currently few effective drugs for cocaine overdose. We have proposed that in patients who have overdosed on cocaine, administration of a recombinant enzyme (e.g. a CE) that could metabolize the drug to inactive metabolites, may be effective in ameliorating the toxicity associated with this agent (Potter and Wadkins, 2006; Redinbo and Potter, 2005). However, for any approach to be successful, a detailed analysis of the metabolism of cocaine by different enzymes will be essential.
Although the metabolism of heroin and cocaine has been presumed to occur via CE-mediated hydrolysis of the ester functions, it is clear that butyrylcholinesterase (BChE) can also hydrolyse these drugs (Gatley, 1990; Lockridge and LaDu, 1980). However, the relative contribution of CEs and BChE towards metabolism of each of these substrates is unclear, in part due to the lack of availability of the purified proteins, and also due to the lack of specific reagents that could inhibit the different enzyme isoforms.
As hCE1 (CES1), hiCE (CES2) and BChE are all expressed in the liver, the determination of which enzyme can metabolize particular substrates has been difficult. In this article, we have purified recombinant hiCE (CES2) to homogeneity, assessed its hydrolytic activity towards several different substrates including heroin and cocaine, and determined the ability of specific inhibitors to modulate enzyme activity. Furthermore, we have correlated these results using human liver and intestinal specimens and corroborated our experiments using computer-assisted molecular modelling. These studies confirm that cocaine is exclusively metabolized by hiCE (CES2) and not by hCE1 (CES1).
Methods
CE activity assay
CE activity was measured by following o-nitrophenyl acetate (o-NPA) hydrolysis at 420 nm as previously described (Beaufay et al., 1974; Danks et al., 1998). Protein concentrations were determined using the Bradford method (using Bio-Rad reagent, Hercules, CA, USA). Data were expressed as nmoles of o-nitrophenol produced per minute per milligram of protein (nmol/min/mg) as calculated using the extinction coefficient for o-nitrophenol (13.6 × 103 M−1 cm−1).
Kinetics of nitrophenyl ester hydrolysis
The kinetic parameters Km, kcat and Vmax for a panel of nitrophenyl esters (Table 1) were determined as previously described (Wadkins et al., 2001). Briefly, assays were performed in 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.4 and formation of the products was determined spectrophotometrically. Product yields were then calculated using the extinction coefficients for nitrophenol, and kinetic parameters were determined from these values
Table 1.
Purification of hiCE (CES2) to homogeneity
| Sample | Volume (mL) | Total activity (U) | Total protein (mg) | Specific activity (U mg−1) | Recovery (%) |
|---|---|---|---|---|---|
| Crude media | 1100 | 472 200 | 300 | 1 550 | 100 |
| After ultrafiltration | 520 | 472 700 | 260 | 1 840 | 100 |
| After IEC | 146 | 116 400 | 4.00 | 29 300 | 25 |
| After ultrafiltration | 0.3 | 96 900 | 3.20 | 30 700 | 21 |
| After Sephacryl S200 | 5 | 41 700 | 0.80 | 52 100 | 9 |
A typical purification profile for hiCE (CES2), isolated from serum-free media, harvested from Spodoptera frugiperda Sf21 cells expressing a secreted form of the enzyme. Carboxylesterase (CE) activity was determined using o-NPA as a substrate and 1 U is equivalent to 1 nmol of o-nitrophenol produced per minute.
Kinetics of CPT-11 metabolism
The kinetic parameters for CPT-11 hydrolysis were determined as previously described (Wadkins et al., 2001).
Analysis of cocaine hydrolysis
To assess the hydrolysis of cocaine to benzoylecgonine and ecgonine methyl ester, the drug (1 mM) was incubated with either media harvested from Sf21 cells (150 µL) or purified CEs (20 µg) in 50 mM HEPES pH 7.4 at 37°C. After 60 min, samples were analysed by high-performance liquid chromatography (HPLC) for metabolite formation. For kinetic studies, 0–5 mM of cocaine was incubated with hiCE (CES2) (250 U/reaction, 10 min) at 37°C. One unit of enzyme activity is equivalent to 1 nmol of nitrophenol produced per min. Reactions were terminated by addition of cold acidified methanol and drug concentrations were determined using isocratic HPLC analysis. Samples (20 µL) were separated using a 3.9 × 300 mm 4 µm Nova-Pak C18 reverse-phase column (Waters Corporation, Milford, MA, USA) and a mobile phase containing 25% (v/v) acetonitrile in 50 mM NH4H2PO4, 150 µM tetrabutyl ammonium phosphate (TBAP), pH 2.2. Metabolites were eluted at a flow rate of 1.0 ml/min, and identified by ultraviolet detection (235 nm). As ecgonine methyl ester absorbs poorly at this wavelength, hydrolysis of cocaine to this product was monitored by the formation of benzoic acid (the acid released from enzymatic hydrolysis). Under these conditions, cocaine, benzoylecgonine and benzoic acid eluted at 4.3, 2.8 and 7.4 min, respectively. Drug concentrations were calculated using System Gold software (Beckmann, Brea, CA, USA) by comparison with known amounts of standard compounds. The sensitivity of detection for this system was 2, 2 and 1 ng/µL for cocaine, benzoylecgonine and benzoic acid, respectively. All reactions were corrected for non-enzymatic hydrolysis by subtraction of the levels of products when cocaine was incubated with buffer alone.
Analysis of heroin hydrolysis
Enzymatic hydrolysis of heroin to 6-monoacetylmorphine was examined under the following conditions. Drug was added to a pre-warmed (37°C) mixture containing purified enzyme and 50 mM HEPES buffer pH 7.4. Typically, 0–5 mM heroin was incubated for 2 min with hCE1 (CES1) (25 U/reaction) or hiCE (CES2) (10 U/reaction), at 37°C. All reactions were terminated by addition of equal volume of ice-cold acidified methanol (containing 0.5% 1 M HCl), and proteins were pelleted by centrifugation for 5 min at 14 500×g at 4°C.
The supernatants from the above reactions were then analysed by reverse phase HPLC using a similar system to that for cocaine. However, heroin and its metabolites were separated and quantitated using a gradient profile as follows: 100% buffer A for 1.5 min changing to 100% buffer B over 0.5 min for 5.5 min [where buffer A is 5% (v/v) acetonitrile in 50 mM NH4H2PO4, 150 µM TBAP, pH 2.2 and buffer B is 25% (v/v) acetonitrile in 50 mM NH4H2PO4, 150 µM TBAP, pH 2.2]. Using a flow rate of 1 ml/min, heroin, 6-acetylmorphine and morphine eluted at 7.0, 5.6 and 4.1 min, respectively, and were detected at 235 nm. Concentrations were determined in an identical fashion to that described for cocaine earlier, and the sensitivity of this system was 3 ng·µL−1, 2 ng·µL−1 and 4 ng·µL−1 for heroin, 6-acetylmorphine and morphine, respectively. All reactions were corrected for non-enzymatic hydrolysis by subtraction of the levels of products when cocaine was incubated with buffer alone.
Prediction of logP values
Calculated logP (clogP) values were obtained using ChemSilico Predict v2.0 software (ChemSilico LLC, Tewksbury, MA, USA).
Experimental design
Expression and purification of recombinant hiCE (CES2)
The purification of hiCE (CES2) was achieved by overexpression of a secreted form of the protein from Spodoptera frugiperda Sf21 cells using baculoviral expression vectors (Morton and Potter, 2000). All purification steps were carried out at 4°C. Media harvested from infected Sf21 cells was buffer exchanged with 50 mM HEPES pH 7.4 buffer using ultrafiltration with Ultracel PL-30 membranes (Millipore, Bedford, MA) and the sample was then applied to a 2.5 × 6 cm macro-prep diethylaminoethyl (DEAE) column equilibrated in the same buffer. Following washing with 1 column volume (∼30 mL) of 50 mM HEPES pH 7.4, the sample was then washed with 5 column volumes of 50 mM HEPES pH 7.4 buffer containing 125 mM NaCl. Elution of hiCE (CES2) was accomplished with 50 mM HEPES pH 5.5 buffer containing 130 mM NaCl. The hiCE (CES2)-containing fractions were pooled, concentrated by ultrafiltration and applied to a 1.5 × 170 cm Sephacryl S-200 HR column (column volume ∼300 mL; Amersham Biosciences Inc., Piscataway, NJ, USA), pre-equilibrated with 50 mM HEPES pH 7.4. hiCE (CES2) was eluted from the column using a flow rate of 6 mL·h−1 and was estimated to be greater than 98% pure by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE).
Chromatography of human intestinal microsomal extracts
Extracts, typically containing ∼10 mg of total protein (equivalent to ∼10 000 U of CE activity), were applied to a 1 × 170 cm Sephacryl S-200 HR column equilibrated in 150 mM NaCl, 50 mM HEPES pH7.4, 0.5% Triton X-100 and samples were eluted using the same buffer. Routinely 3 ml fractions were collected, concentrated and then assayed for the desired enzyme activity. Western analysis for hCE1 (CES1) and hiCE (CES2) was undertaken using specific antibodies for these proteins using chemiluminescent detection.
Data analysis and statistical procedures
Enzyme inhibition assays
CE enzyme inhibition was determined using the previously described, selective CE inhibitors (Wadkins et al., 2004; Wadkins et al., 2005). Compounds analysed were benzil (1), 1,4-bisbenzil (2), 1-(3,4-dimethyl-phenyl)-2-phenyl-ethane-1,2-dione (3), N,N'-(2,3,5,6-tetramethyl-1,4-phenylene)bis(4-fluorobenzene sulfonamide) (4) or N,N'-1,4-phenylenebis(3-bromobenzene sulfonamide) (5) (see the chemical structures of these compounds in Table 4) using cocaine, heroin, o-NPA or CPT-11 as substrates. Briefly, the formation of product in the presence of inhibitor at concentrations ranging from 1 nM to 100 µM was determined (Wadkins et al., 2004; Wadkins et al., 2005). Routinely at least eight concentrations of inhibitor were used. To determine the Ki values (inhibition constants), data were fitted to the following equation (Webb, 1963):
Table 4.
Ki values for the inhibition of hiCE (CES2) and hCE1 (CES1) by a variety of CE inhibitors
| Inhibitor | Structure |
hiCE (CES2) Ki±s.e.m. (nM) |
hCE1 (CES1) Ki±s.e.m. (nM) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| o-NPA | Cocaine | Heroin | CPT-11 | o-NPA | Cocaine | Heroin | CPT-11 | ||
| 1 | 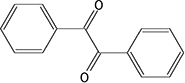 |
14.7 ± 1.9a | 85.5 ± 19.0 | 92.4 ± 12.5 | 40.0 ± 10.0 | 45.1 ± 3.2a | NAb | 45.7 ± 8.8 | NAb |
| 2 | 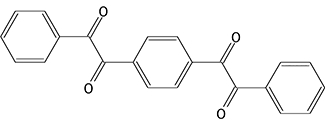 |
5.6 ± 0.4a | 88.9 ± 22.0 | 42.2 ± 7.7 | 13.9 ± 2.3 | 8.0 ± 0.9a | NA | 29.2 ± 9.5 | NA |
| 3 | 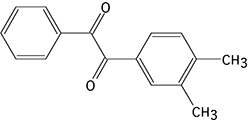 |
4.1 ± 0.4a | 53.3 ± 11.5 | 20.1 ± 5.1 | 13.9 ± 2.3 | 99.1 ± 10.0a | NA | 77.7 ± 16.9 | NA |
| 4 | 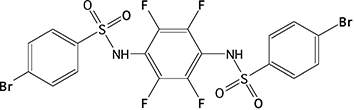 |
23.7 ± 2.7c | 69.5 ± 21.9 | 111 ± 17 | 35.8 ± 8.3 | >100 000 | NA | >100 000 | NA |
| 5 | 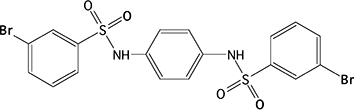 |
67.0 ± 9.6c | 38.7 ± 10.6 | 404 ± 78 | 144 ± 27 | >100 000 | NA | >100 000 | NA |
The inhibition of the hydrolysis of o-NPA, cocaine, heroin or CPT-11 by hiCE (CES2) or hCE1 (CES1), with various CE inhibitors, was determined as described in the text.
Data has been reported previously (Hyatt et al., 2005) and is included for comparative purposes.
NA, not applicable. The Ki values were not determined for these substrates due to their very poor hydrolysis by hCE1 (CES1).
Data has been recently reported (Hicks et al., 2009) and is included for comparative purposes.
s.e.m., standard error of the mean; CE, carboxylesterase.
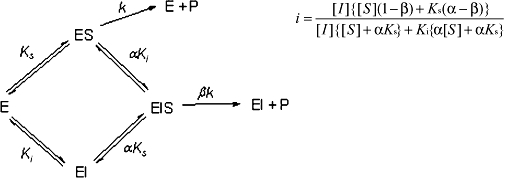 |
where i= fractional inhibition, [I]= inhibitor concentration, [S]= substrate concentration, α= change in affinity of the substrate for the enzyme in the presence of the inhibitor (where α > 0), b= change in the rate of enzyme substrate complex decomposition in the presence of the inhibitor (where 1 > b > 0), Ks is the dissociation constant for the enzyme substrate complex (assuming negligible commitment to catalysis) and Ki is the inhibitor constant. The data sets were analysed using Prism 4.0 software (GraphPad, La Jolla, CA, USA) and Perl Data Language, and the mode of enzyme inhibition was determined by evaluating the r2 values for the curve fits using Akaike's information criteria (Wadkins et al., 2005). Ki values were then calculated using the best fit model described from these analyses.
In most cases, enzyme inhibition was partially competitive (i.e. where the inhibitor does not affect rate of enzyme/substrate complex dissociation and only partially hinders substrate binding).
Molecular modelling studies
Molecular modelling analyses were performed using ICM Pro software v3.6 (Molsoft LLC, La Jolla, CA, USA) running on a dual Xeon computer in a Windows XP environment. Recent studies indicate that docking experiments using ICM Pro compare favorably with FlexX, GOLD and GLIDE (Chen et al., 2006). For hCE1 (CES1), the coordinates derived from the CE/homatropine crystal structure were used (1MX5; Bencharit et al., 2003b). For hiCE (CES2), a homology model was developed using the 1MX5 coordinates With Modeller 8 (Sali and Blundell, 1993) software.
For these analyses, substrate and inhibitor structures were constructed in ChemDraw (CambridgeSoft, Cambridge, MA, USA) and subjected to energy minimization using ICM-Pro. The receptor site, consisting of the residues that encompassed the enzyme active site gorge, was then selected as a pocket into which the minimized ligand could be docked. For cocaine and heroin, binding of both the protonated and the native form of the molecule was assessed. Docking studies were performed using essentially the default parameters for the ICM Pro software. A rigid receptor/flexible ligand approach was adopted that used five potential energy maps combining hydrophobicity, electrostatics, hydrogen bond formation and two van der Waals parameters. However, minor modifications included constructing receptor electrostatic charge maps at a resolution of 0.1 Å and increasing the ‘Thoroughness’ parameter to 10. The latter increased the time and number of poses that were evaluated during the docking simulations.
Materials
Cocaine ((–)-cocaine) and heroin were purchased from Cerilliant Corporation (Round Rock, TX) and were dissolved in acetonitrile. Routinely, solvent concentrations in biochemical reactions did not exceed 2%, although no effect was seen on CE activity at 5% acetonitrile. CPT-11 was kindly provided by Dr J. P. McGovren (Pharmacia, Peapack, NJ, USA) and was dissolved in methanol. The Macro-Prep® DEAE support was from Bio-Rad and Sephacryl S-200 high resolution resin was obtained from Amersham Biosciences Inc. All other chemicals were purchased from Fisher (Fair Lawn, NJ, USA), Organic Research (Cincinnati, OH, USA), Sigma-Aldrich (St. Louis, MO, USA) or VWR Scientific Products, Inc. (Suwanee, GA, USA).
hCE1 (CES1) was purified from the media of baculovirus-infected Spodoptera frugiperda Sf21 cells as previously described (Bencharit et al., 2003a; Bencharit et al., 2003b; Morton and Potter, 2000). The GenBank accession numbers for the cDNAs encoding the CEs used in these studies were as follows; hiCE (CES2) - Y09616 (Schwer et al., 1997); and hCE1 (CES1) – M73499 (Munger et al., 1991). Rabbit polyclonal antibodies directed towards hiCE (CES2) and hCE1 (CES1) were generated by repeated immunization of rabbits with recombinant proteins. Antibodies were purified from sera using IgA affinity chromatography and sensitivity and specificity were verified by enzyme-linked immunosorbent assay and Western analyses. Human liver and intestinal microsomal extracts were purchased from BD Biosciences (Woburn, MA, USA) and Celsis In Vitro (Baltimore, MD, USA), respectively.
Results
Expression and purification of recombinant hiCE (CES2)
Media was obtained from S. frugiperda Sf21 cells that had been infected with baculovirus expressing a secreted form of the hiCE (CES2) protein (Potter et al., 1998; Khanna et al., 2000). Initial attempts at purification using isoelectric focusing (Morton and Potter, 2000) proved unsuccessful and ion-exchange chromatography was chosen as the preferred method. A step-wise wash and elution was used to allow the separation of hiCE (CES2) from two problematic contaminants, bovine serum albumin (present in minor amounts in the culture media) and baculoviral chitinase, both of which have molecular masses similar to hiCE (CES2) (∼65 kDa). The anion-exchange column allowed for a 15-fold purification followed by an additional 2-fold purification by gel filtration, based on yields of CE activity (Table 1). Typically, we obtained 1 mg of hiCE (CES2) per liter of insect cell culture media. Multiple batches of hiCE (CES2) have been made by this protocol with final specific activities ranging from 40 to 55 000 U·mg−1, with a typical average of 50 000 U·mg−1; however, selected fractions typically showed a specific activity as high as 60 000 U·mg−1. Purity of the samples was assessed by SDS-PAGE analysis (Figure 1) and protein identification by Matrix-assisted laser desorption/ionization-time of flight time of flight mass spectrometry.
Figure 1.
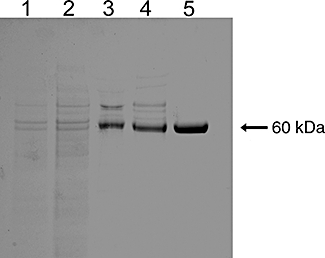
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis analysis of different fractions following purification of hiCE (CES2). Approximately 3 µg of protein was loaded into each lane and following electrophoresis, the gel was stained with Coomassie Blue. Lane 1, Crude culture media; Lane 2, After ultrafiltration; Lane 3, After ion-exchange chromatography using DEAE; Lane 4, After ultrafiltration; Lane 5, After chromatography on Sephacryl S200 HR.
To determine the oligomerization state of hiCE (CES2), we calibrated a Sephacryl S200 column using low molecular mass protein standards and applied a sample of the purified CE. These studies indicated that hiCE (CES2) had a predicted molecular mass of ∼66 kDa. Furthermore, in co-chromatographic studies, it was observed that hiCE (CES2) and bovine serum albumin (66 kDa) eluted together off this Sephacryl column. As the hiCE (CES2) cDNA contains an open reading frame that would give rise to a protein of predicted molecular mass of 61 807 Da (Schwer et al., 1997), we conclude that under the conditions employed here, the hiCE (CES2) protein purifies as a monomer.
Kinetic parameters of recombinant hiCE (CES2) with nitrophenyl ester substrates
Having isolated recombinant hiCE (CES2) and having homogenous hCE1 (CES1) at hand (Bencharit et al., 2003b; Morton and Potter, 2000), we evaluated the kinetic parameters of these proteins with a variety of different esterase substrates. The Km, Vmax and kcat/Km were therefore determined for various nitrophenyl esters using hiCE (CES2), and were compared with those for hCE1 (CES1) (Table 2). As can be seen, the Km values demonstrated considerable variation with no obvious correlation with either enzyme. However, the Vmax values for hiCE (CES2) ranged from 0.77 to 12.0 nmol·min−1 for the different substrates, and these values were up to 6-fold higher than that observed for hCE1 (CES1). Peak velocities were seen with p-nitrophenyl butyrate and p-nitrophenyl propionate for hiCE (CES2) and hCE1 (CES1), respectively. However, maximal catalytic efficiency for hCE1 (CES1) was demonstrated with the valerate ester. This is a consequence of the much lower Km constants that are observed with this substrate, resulting in higher kcat/Km values.
Table 2.
A comparison of kinetic parameters of hiCE (CES2) and hCE1 (CES1) with various nitrophenyl esters
| Substrate | clogP | Enzyme | Km±s.e.m. (µM) | Vmax±s.e.m. (nmol min−1) | kcat/Km (s−1 mM−1) | Ratio of kcat/Km[hiCE (CES2)/ hCE1 CES1] |
|---|---|---|---|---|---|---|
| o-Nitrophenyl acetate | 1.12 | hiCE (CES2) | 79 ± 7.6 | 0.94 ± 0.02 | 67 ± 6 | 8.5 |
| hCE1 (CES1) | 73 ± 7.4 | 0.15 ± 0.00 | 7.9 ± 0.7 | |||
| p-Nitrophenyl acetate | 1.16 | hiCE (CES2) | 976 ± 60 | 4.17 ± 0.14 | 24 ± 1 | 6.3 |
| hCE1 (CES1) | 822 ± 73 | 0.81 ± 0.02 | 3.8 ± 0.3 | |||
| p-Nitrophenyl propionate | 1.86 | hiCE (CES2) | 201 ± 18 | 9.3 ± 0.6 | 261 ± 7 | 8.2 |
| hCE1 (CES1) | 249 ± 9 | 2.05 ± 0.03 | 32 ± 0.6 | |||
| p-Nitrophenyl butyrate | 2.46 | hiCE (CES2) | 117 ± 5 | 12 ± 0.4 | 580 ± 5 | 15.7 |
| hCE1 (CES1) | 169 ± 8 | 1.65 ± 0.04 | 37 ± 1 | |||
| p-Nitrophenyl trimethylacetate | 2.75 | hiCE (CES2) | 19.8 ± 1.1 | 0.77 ± 0.02 | 220 ± 9 | 14.7 |
| hCE1 (CES1) | 29 ± 5 | 0.8 ± 0.1 | 15 ± 0.6 | |||
| p-Nitrophenyl valerate | 3.01 | hiCE (CES2) | 71 ± 11 | 10.5 ± 1.2 | 421 ± 27 | 8.1 |
| hCE1 (CES1) | 27 ± 2.7 | 0.73 ± 0.04 | 52 ± 2.5 |
Assessment of substrate hydrolysis was determined spectrophotometrically by monitoring the formation of nitrophenol at 420 nm. Kinetic parameters were then calculated from these data sets.
s.e.m., standard error of the mean; CE, carboxylesterase.
It was also noted that with the larger, more bulky substrates, such as p-nitrophenyl butyrate or p-nitrophenyl trimethylacetate, hiCE (CES2) was much more proficient at substrate hydrolysis. This is exemplified by the significantly larger kcat/Km values for these esters (Table 2). For example, hiCE (CES2) was ∼16-fold more efficient at p-nitrophenyl butyrate hydrolysis than hCE1 (CES1). As indicated in Table 2, for all substrates analysed, hiCE (CES2) was more efficient at substrate turnover as compared with hCE1 (CES1).
As the active sites of the CEs are lined with aromatic amino acids (Bencharit et al., 2003b; Potter and Wadkins, 2006), we hypothesized that more hydrophobic molecules would preferentially localize within these domains. Therefore, we compared the affinity constants (Km) for the para- series of substituted nitrophenyl esters for the two CEs, with their clogP values (calculated water/octanol partition coefficients). As shown in Figure 2, excellent correlations were seen with data obtained from both enzymes, with r2 for the curve fits of 0.79 and 0.89 for hiCE (CES2) and hCE1 (CES1), respectively. These results confirm that the affinity of the compounds for the CEs is directly related to their clogP (or hydrophobicity). This is consistent with the notion that the longer alkyl chain esters localize within the hydrophobic active site gorges present within the protein.
Figure 2.
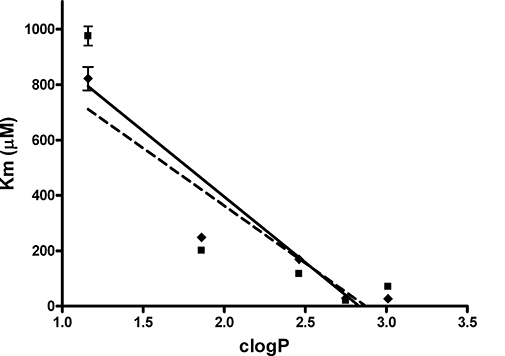
A graph demonstrating the linear relationships between the clogP values of the para-substituted nitrophenyl esters, and their observed Km constants for hiCE (CES2) ( ), and hCE1 (CES1) (
), and hCE1 (CES1) ( ). The correlation coefficients (r2) for the data sets are indicated on the graph.
). The correlation coefficients (r2) for the data sets are indicated on the graph.
Metabolism of cocaine by CEs
Having demonstrated that hiCE (CES2) could hydrolyse large, bulky esters, we evaluated the ability of this enzyme to metabolize cocaine and heroin. As before, we also determined the ability of recombinant hCE1 (CES1) to hydrolyse these compounds. As indicated in Table 3 and displayed in Figure 3, hiCE (CES2) readily hydrolysed cocaine to yield benzoic acid (and ecgonine methyl ester) with Km and Vmax values of 202 µM and 589 pmol·min−1, respectively. In contrast, no hydrolysis of the drug to either benzoylecgonine, or benzoic acid, was observed with hCE1 (CES1) (Figure 3A). Indeed, no enzyme-mediated metabolism of cocaine was seen, even after prolonged incubation (∼20 h) with hCE1 (CES1) (data not shown). Under these same conditions using hiCE (CES2), all of the cocaine was hydrolysed to benzoic acid (data not shown). We conclude therefore, that hiCE (CES2) is the primary CE responsible for cocaine metabolism in vivo.
Table 3.
Kinetic parameters for the metabolism of heroin, cocaine, and CPT-11 for hiCE (CES2) and hCE1 (CES1)
| Substrate | Enzyme | Km±s.e.m. (µM) | Vmax±s.e.m. (pmol min−1) | kcat/Km (s−1 mM−1) | Ratio of kcat/Km hiCE (CES2)/hCE1 (CES1) |
|---|---|---|---|---|---|
| Heroin | hiCE (CES2) | 1440 ± 630 | 8130 ± 910 | 5.9 | 0.65 |
| hCE1 (CES1) | 445 ± 73 | 10 660 ± 450 | 9.7 | ||
| Cocaine | hiCE (CES2) | 202 ± 45 | 589 ± 33 | 0.44 | – |
| hCE1 (CES1) | NDa | NDa | - | ||
| CPT-11 | hiCE (CES2) | 3.9 ± 0.3 | 1.6 ± 0.12 | 0.41 | 93 |
| hCE1 (CES1) | 82.8 ± 9.6 | 0.36 ± 0.02 | 0.0044 |
The hydrolysis of the respective substrates was determined using HPLC separation and detection of the products, from which the kinetic parameters were calculated.
ND, not determined. No hydrolysis was apparent for this substrate.
s.e.m., standard error of the mean; CE, carboxylesterase.
Figure 3.
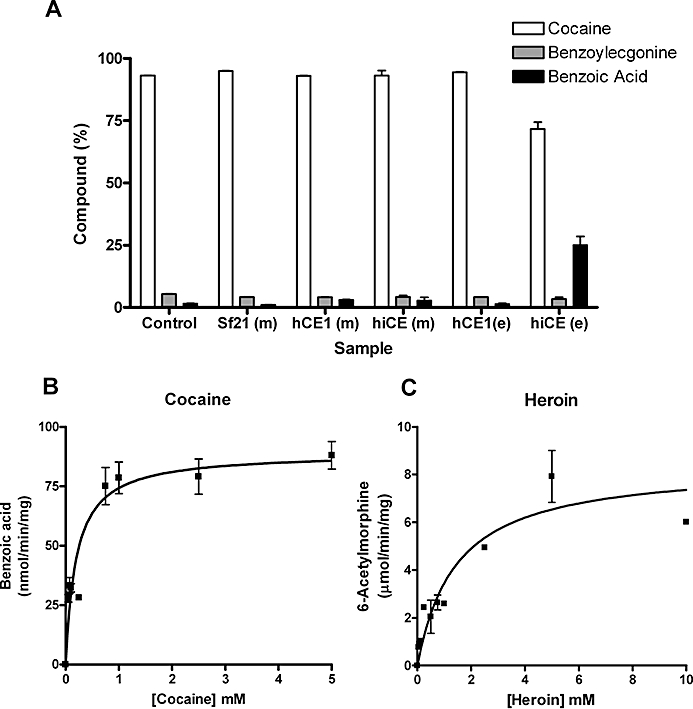
(A) Metabolism of cocaine by different carboxylesterases (CEs). Cocaine (1 mM) was incubated with the different samples for 1 h. The yields of benzoylecgonine and benzoic acid were determined by HPLC. Essentially, only hiCE (CES2) was able to hydrolyse cocaine. Control – 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer pH 7.4 only; Sf21 (m) – media harvested from uninfected Sf21 cells; hCE1 (m) – media harvested from Sf21 cells infected with baculovirus expressing hCE1 (CES1) (∼250 units of CE); hiCE (m) – media harvested from Sf21 cells infected with baculovirus expressing hiCE (CES2) (∼75 units of CE); hCE1 (e) – reaction containing pure hCE1 (CES1) (20 µg =∼2000 units of CE); hiCE (e) – reaction containing pure hiCE (CES2) (20 µg =∼500 units of CE). (B) The concentration versus velocity curve for hiCE (CES2) with cocaine. No evidence of enzyme inhibition by high concentrations of substrate was observed in these assays. (C) The concentration versus velocity curve for hiCE (CES2) with heroin. Similar to that seen for cocaine, no substrate-mediated enzyme inhibition was observed.
Analysis of the curve fits for benzoic acid formation versus substrate concentration (Figure 3B), indicated that no inhibition of hiCE (CES2) activity was observed with high concentrations of cocaine (up to 5 mM). This is in contrast to that seen for BChE where cocaine concentrations greater than 300 µM resulted in reduced product formation (Stewart et al., 1977). This suggests that in situations where individuals overdose with very high levels of drug, hiCE (CES2) may play a significant role in detoxification of cocaine.
The metabolism of the anticancer agent CPT-11 was included in these studies since previous reports have indicated that this substrate is hydrolysed much more effectively by hiCE (CES2) than by hCE1 (CES1) (Humerickhouse et al., 2000). Our studies are in agreement with these results (Table 3), confirming the specificity and hydrolytic activity of the recombinant purified hiCE (CES2) protein.
Metabolism of heroin by CEs
In contrast to the results obtained when using cocaine as a substrate, both human CEs did hydrolyse heroin. The efficiencies of hydrolysis of this drug to 6-acetylmorphine were similar, as exemplified by the kcat/Km values (Table 3). It should be noted however, that the kcat/Km values for heroin catalysis [5.9 and 9.7 s−1 mM−1 for hiCE (CES2) and hCE1 (CES1), respectively] were at least 10-fold higher than that observed for cocaine with hiCE (CES2) (0.44 s−1 mM−1). Indeed, based upon these kinetic analyses, the catalytic efficiency of heroin hydrolysis by CEs was ∼12- to 40-fold more efficient than cocaine metabolism by the latter enzyme. Again, no inhibition of hiCE (CES2) enzyme activity was observed in the presence of high concentrations of heroin (Figure 3C).
Inhibition of hiCE (CES2) and hCE1 (CES1) with CE-specific inhibitors
As the inhibition of CE-mediated hydrolysis of drugs may be used to modulate the levels of toxic metabolites (such as morphine from heroin), we determined the ability of a series of recently identified CE inhibitors to alter heroin, cocaine or CPT-11 hydrolysis. The benzil based compounds (1-3) are generic CE inhibitors, whereas the bis-benzene sulfonamides, 4 and 5, demonstrate specificity for hiCE (CES2). As indicated in Table 4, inhibitors 1-3 displayed very low Ki values towards hiCE (CES2) and hCE1 (CES1) for all substrates. With compounds 4 and 5, which are hiCE (CES2)-specific inhibitors (Wadkins et al., 2004), no inhibition of metabolism of o-NPA or heroin was observed with hCE1 (CES1). However, potent inhibition of hiCE (CES2)-mediated metabolism was seen for all substrates, with Ki values as low as 35 nM with heroin. These studies confirm the applicability of using inhibitors to modulate these reactions and indicate that the selectivity of the sulfonamides towards hiCE (CES2) may have utility in modulating either CPT-11 or heroin hydrolysis in vivo.
Metabolism of heroin and cocaine by human intestinal and liver extracts
To confirm the results observed with the recombinant protein, we evaluated the ability of human intestinal and liver microsomal extracts to hydrolyse the drugs. We chose these tissues because they are rich in hiCE (CES2) and hCE1 (CES1), respectively, and demonstrate significantly different ratios of the two human CEs. This is exemplified by the Western analysis of total microsomal extracts shown in Figure 4A and the hydrolytic activities of these samples (Figure 4B). As can be seen, the intestinal microsomes contained significantly more hiCE (CES2) than hCE1 (CES1); the converse being true for the liver sample.
Figure 4.
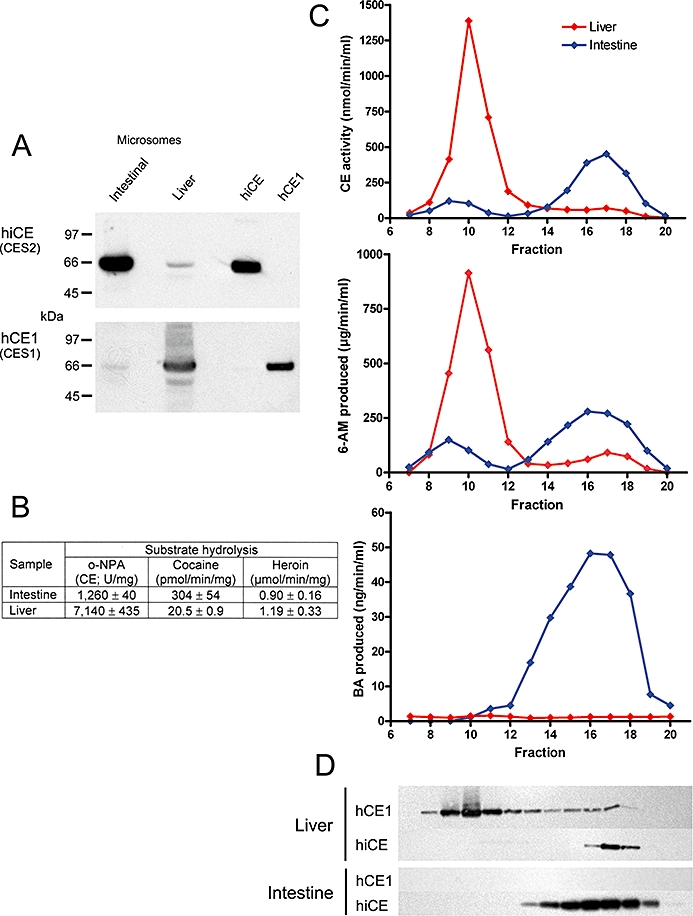
Chromatographic separation of carboxylesterases (CEs) in human intestinal and liver microsomal extracts using Sephacryl S-200 HR resin. (A) Western analysis demonstrating the amounts of hiCE (CES2) and hCE1 (CES1) present in the samples prior to chromatography. Standards present on the right hand side of the image represent 50 ng of each pure protein. (B) Table demonstrating the levels of CE activity, as well as the cocaine and heroin hydrolysis of the intestine and liver microsomal extracts prior to chromatography. (C) Elution profile for intestinal (blue line) and liver (red line) microsomal extracts demonstrating levels of CE activity (top graph), 6-acetylmorphine (6-AM) formation (heroin hydrolysis; middle graph) and benzoic acid (BA) production (cocaine hydrolysis; bottom graph). (D) Western analysis of these same fractions analysed in panel C, using antibodies specific for hiCE (CES2) or hCE1 (CES1). The images are aligned such that the signals correspond to the fraction number indicated on the abscissa axis.
Chromatography of extracts was performed on Sephacryl S-200 HR and the CE activity of individual fractions was determined. In addition, the presence of hiCE (CES2) or hCE1 (CES1) in each fraction was assessed by Western analysis and hydrolysis of cocaine and heroin was monitored by HPLC. Figure 4C, D demonstrate the activity profile of the fractions obtained from this chromatographic technique.
As can be seen with the intestinal microsomal sample, two peaks of esterase activity were observed (fractions 8–11 and 14–19) with the latter correlating with the presence of hiCE (CES2) (see the Western analyses under the graph –Figure 4D). Interestingly, the earlier peak did not correspond to hCE1 (CES1) or BChE (data not shown), suggesting that this represents another unknown esterase in this sample. Assessment of cocaine and heroin metabolism indicated that although the latter was effectively converted to 6-acetylmorphine by both fractions, only hiCE (CES2) was capable of hydrolyzing the former drug (Figure 4C, bottom graph). Although the identity of this enzyme that eluted between fractions 8–11 is not known, the data presented in Figure 4 indicate that this protein did not contribute to cocaine hydrolysis.
As a comparison, we performed similar studies using a human liver microsomal extracts that demonstrated high expression of hCE1 (CES1) and low levels of hiCE (CES2) (see Figure 4A). Little or no cocaine hydrolysis was observed with either the initial extract (Table in Figure 4B) or from any of the fractions following chromatography (Figure 4C, bottom graph, red line), even though large amounts of hCE1 (CES1) were present in these samples (Figure 4D). Although hiCE (CES2) was present in the liver sample, the actual level of this enzyme was very low (see Figure 4A) and the cocaine hydrolysis products were probably below the limit of detection for these assays.
Overall, the results from these studies are consistent with the in vitro experiments using recombinant protein, that demonstrate that hiCE (CES2) is responsible for conversion of cocaine to benzoic acid and ecgonine methyl ester.
Molecular analysis of cocaine and heroin binding to CEs
Having demonstrated that hiCE (CES2) was markedly more efficient at cocaine hydrolysis than hCE1 (CES1), we sought to determine whether molecular modelling studies could provide insight into the alignment of the substrates within the enzyme active sites. Therefore, we docked the drug (and a variety of other compounds) into the active sites of both enzymes. As the crystal structure for hiCE (CES2) is currently not available, we generated a homology model using the coordinates for hCE1 (CES1) when bound to homatropine.
Interestingly, when the hCE1 (CES1) and derived hiCE (CES2) structures were overlaid, the catalytic residues (Ser, His and Glu) were essentially in the same position and orientation in both proteins (Figure 5B). However, as can be seen in Figure 5A, one loop of amino acids (residues 303-320 in hCE1) was displaced as compared with hiCE (CES2). It is unclear whether movement of this domain affects either the biological activity and/or docking studies, but due its proximity to the active site, it is likely to play a role in substrate hydrolysis.
Figure 5.
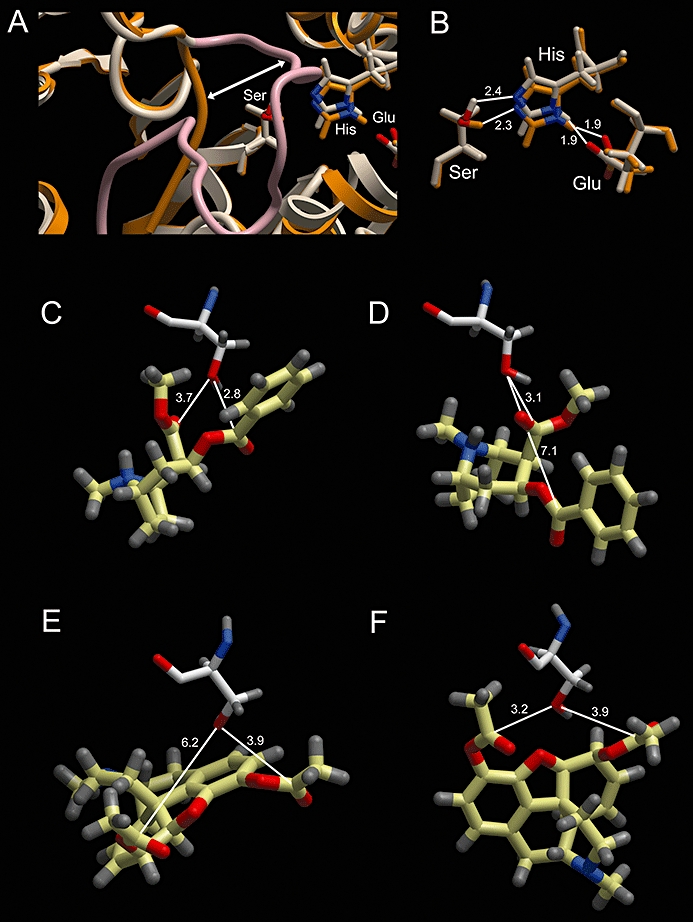
Computer-assisted docking of cocaine or heroin into the active sites of hCE1 (CES1) or hiCE (CES2). (A) An overlay of the ribbon representations of the hCE1 (CES1) (taupe) and hiCE (CES2) (orange/brown) structures used for the docking studies. As can be seen one loop of hCE1 (CES1) (highlighted in pink) is displaced (marked by the white arrow) and projects towards the active site catalytic amino acids (Ser, His, Glu). (B) An overlay of the catalytic amino acids in hCE1 (CES1) (taupe) and hiCE (CES2) (orange/brown) with the distances between the relevant atoms indicated. (C, D) – Docking of cocaine into the active sites of hiCE (CES2) and hCE1 (CES1), respectively. The active site serine is depicted and the distance from the Oγ atom to the carbonyl carbon atoms in the drug are displayed. (E, F) – Docking of heroin into the active sites of hiCE (CES2) and hCE1 (CES1), respectively. The active site serine is depicted and the distance from the Oγ atom to the carbonyl carbon atoms in the drug are displayed. In all panels, the nitrogen and oxygen atoms displayed in blue and red, respectively, and distances are marked in Å.
Following docking of different molecules the distances between the catalytic serine residue and the carbonyl carbon atoms were measured. As can be seen in Table 5 and Figure 5, the carbonyl carbon atom of the benzyl ester in cocaine localized 2.8 Å from the serine Oγ atom for hiCE (CES2) (Figure 5C), but was localized greater than 7 Å away in hCE1 (CES1) (Figure 5D). As the latter distance is too large for nucleophilic attack, this suggests that hCE1 (CES1) would be unable to hydrolyse this ester function. This was true for both the protonated and un-protonated forms of the drug.
Table 5.
Distance between the catalytic serine Oγ atom and the carbonyl carbon atoms for a series carboxylesterase (CE) substrates and inhibitors
| Compound |
Distance from Ser OγH to C=O(Å) |
|
|---|---|---|
| hCE1 (CES1) | hiCE (CES2) | |
| Cocainea | 4.9 (Bz) | 2.6 (Bz) |
| 3.9 (Me) | 5.1 (Me) | |
| Cocainea (protonated) | 7.1 (Bz) | 2.8 (Bz) |
| 3.1 (Me) | 3.7 (Me) | |
| Heroinb | 3.2 (3-) | 3.9 (3-) |
| 3.9 (6-) | 6.2 (6-) | |
| Heroinb (protonated) | 2.8 (3-) | 3.6 (3-) |
| 5.7 (6-) | 5.2 (6-) | |
| o-NPA | 3.3 | 3.7 |
| o-Nitrophenyl valerate | 3.7 | 2.8 |
| CPT-11 | NAc | 3.1 |
| Benzild | 3.9, 4.8 | 3.0, 3.3 |
| Camphorquinonee | 5.8 (2-) | 6.7 (2-) |
| 5.0 (3-) | 6.8 (3-) | |
The distance between the catalytic serine Oγ atom and the carbonyl carbon atoms for the listed compounds were determined following docking into the active sites of either hCE1 (CES1) or hiCE (CES2). Values were calculated using ICM Pro software.
Distances are indicated to the carbonyl carbon atom of either the benzyl ester (Bz) or the methyl ester (Me).
Distances are indicated to the carbonyl carbon atom of the methyl ester at either the 3- or 6- position of the molecule.
Docking of CPT-11 into the active site of hCE1 (CES1) was not undertaken as steric constraints present at the entrance to the active site gorge significantly impede access of the drug to the catalytic amino acids (Wadkins et al., 2001; Wierdl et al., 2008).
Two distance values are given for benzil as it contains two carbonyl groups, however, because this molecule is symmetrical, no assignment of these atoms is included.
Distances are indicated to the carbonyl carbon atom present at the 2- or 3-position of the molecule.
However, following docking of heroin, the carbonyl carbon atoms for the methyl ester in the 3-position localized within 3.9 Å for both CEs (Figure 5E and F).
Indeed, for hCE1 (CES1) the distance to the ester was smaller than that for hiCE (CES2). This is in agreement with our experimental results that demonstrate that the kcat/Km value for heroin with hCE1 (CES1) is ∼threefold greater than that seen for hiCE (CES2) Table 3).
As controls for the docking studies, we also assessed the binding of a series of related substrates and inhibitors. As noted in Table 5, the carbonyl carbon atom was present within 3.7 Å of the serine Oγ atom for o-NPA, p-nitrophenyl valerate, CPT-11 and benzil, for both hCE1 (CES1)and hiCE (CES2). Finally as a negative control, we docked camphorquinone into the enzyme active sites. This molecule contains the ethane-1,2-dione chemotype (similar to benzil), but is not an inhibitor of either of the human CEs (Hyatt et al., 2007). As indicated, the measured distances [∼6.8 Å for hiCE (CES2) and 5.0–5.8 Å for hCE1 CES1)] would be too large for nucleophilic attack by the Oγ atom towards this molecule. In summary, the results of these docking studies are in complete agreement with the obtained experimental data.
Discussion and conclusions
We have purified recombinant human intestinal CE, hiCE (CES2), to homogeneity and determined the kinetic parameters for this enzyme with a variety of clinically relevant substrates. This has demonstrated that hiCE (CES2) efficiently metabolized CPT-11, cocaine and heroin. In contrast, the human liver CE, hCE1 (CES1), only hydrolysed heroin. Until recently, the enzymes involved in the metabolism of all of these agents have not been unambiguously identified. For example, although hiCE (CES2) has been proposed as the principal CE responsible for CPT-11 activation in vivo (Humerickhouse et al., 2000; Khanna et al., 2000), other esterases, for example, BChE can also activate this drug (Dodds and Rivory, 1999; Morton et al., 1999). Similarly, previous reports have indicated that cocaine can be metabolized by both hCE1 (CES1)and hiCE (CES2) (Kamendulis et al., 1996; Brzezinski et al., 1997; Pindel et al., 1997). However, these studies were performed using enzymes purified from extracts derived from human liver. As hCE1 (CES1) and hiCE (CES2) (as well as BChE and other esterases) are expressed in this organ, potentially, contamination of ‘pure’ CEs with other liver esterases could confuse the results obtained from these biochemical experiments. We are unable to assess the level of purity of the enzymes used in these previous studies and hence we cannot comment on which protein would be definitively responsible for cocaine hydrolysis; however, it should be noted that no specific assays or techniques were used by these authors to assess the levels of other esterases present in their enzyme preparations.
We relied on the use of recombinant hCE1 (CES1) or hiCE (CES2), purified from the culture media of the insect cell line, S. frugiperda. We have used this approach, rather than purifying the proteins from cell extracts, to avoid any possible cross contamination with intracellular insect CEs, and to minimize proteolytic degradation of the mammalian enzymes. As the only protein contaminants we have observed during the hiCE (CES2) purification have been bovine serum albumin and Autographa californica chitinase, and neither of these proteins has esterase activity, it is likely that the enzymes used in our studies were homogenous. We did not observe hydrolysis of cocaine by the recombinant hCE1 (CES1) that we used in these studies, and as this protein is catalytically active towards a variety of substrates, we cannot confirm the results generated in previous reports that indicate that the drug is metabolized by this enzyme (Kamendulis et al., 1996; Brzezinski et al., 1997).
Potentially, there may be different isoforms of hCE1 (CES1) that result from nucleotide polymorphisms (Zhu et al., 2008) that could account for the discrepancy between our results and those of Brzezinski et al. (1997). However, as we have sequenced the cDNA that encodes the hCE1 (CES1) protein used in these studies (Danks et al., 1999), determined that this enzyme is pure by both physical and biochemical methods (Bencharit et al., 2003b; Morton and Potter, 2000; Wadkins et al., 2001), and obtained the crystal structure of this protein in complex with a variety of different small molecules (Bencharit et al., 2003b; Fleming et al., 2005; Bencharit et al., 2006), our results clearly indicate that the protein encoded by this cDNA cannot hydrolyse cocaine. Additionally, our studies implicate hiCE (CES2) as the principal CE involved in drug hydrolysis. As hiCE (CES2) is also expressed at relatively high levels in liver, skeletal muscle, heart, colon and kidney (Wu et al., 2003), it is likely that this enzyme will be important in the hydrolysis of this molecule, regardless of the route of cocaine administration.
To validate the in vitro studies, we evaluated the esterase levels and drug hydrolyzing properties of intestinal and liver microsomal extracts after chromatographic separation. Consistent with our previous results, we determined that hiCE (CES2) was responsible for cocaine metabolism and that hCE1 (CES1) was inactive towards this substrate (Figure 4). This was achieved using samples that were either, rich in hiCE (CES2) and low in hCE1 (CES1) (intestine), or where the converse was true (liver). Confirmation of the identity of the enzymes involved was documented using antibodies specific for these proteins. Hence, our results using these human microsomal samples corroborate our previous studies using the recombinant proteins.
In an attempt to understand the differences in cocaine hydrolysis by these two CEs, we undertook molecular docking studies using the hCE1 (CES1) crystal structure and a homology model for hiCE (CES2) based upon these same coordinates. Initially, we were concerned that as the former enzyme cannot hydrolyse cocaine, the use of the hCE1 (CES1) coordinates might result in a conformation in hiCE (CES2) that would preclude informative results from these analyses. However, upon docking of this drug (and others small molecules displayed in Table 5), it was apparent that the homology model demonstrated sufficient variation in the active site gorge, such that different conformations of the CE/drug complexes were observed. Gratifyingly, results from these docking studies placed the active site serine Oγ atom within 3.9 Å of the carbonyl carbon atoms for all substrates and inhibitors with all enzymes, the exceptions being hCE1 (CES1) with cocaine and camphorquinone with both enzymes. The latter 1,2-dione is not an inhibitor of these CEs (Hyatt et al., 2007), even though it demonstrates properties suitable for enzyme inhibition. The lack of CE inhibition and the failure of camphorquinone to dock adjacent to the catalytic residues is likely to be due to steric hindrance enforced by the methyl bridge present in this bicyclic structure (Hyatt et al., 2007).
In previous modelling studies, based upon the crystal structure of homatropine bound in the hCE1 (CES1) active site (Bencharit et al., 2003b), the tropine ring of cocaine was overlaid onto the coordinates for the corresponding moiety of the former molecule. This demonstrated that (–)-cocaine would be preferentially hydrolysed by hCE1 (CES1). However, in these experiments, no computational docking studies were performed and hence the electrostatics of drug binding were not taken into account. Here, we demonstrate that when these forces are considered in the context of cocaine docking into the active sites of the CEs, significant differences in the interaction of the drug with the respective catalytic residues were observed. This resulted in models that confirmed our experimental data, that is, that the carbonyl group present within the benzyl ester chemotype of cocaine was juxtaposed to the catalytic serine Oγ atom in hiCE (CES2) (2.8 Å), but was up to 4 Å further away in hCE1 (CES1). This would make drug hydrolysis by the latter enzyme unlikely, again consistent with the biochemical results presented earlier.
Recent analysis of the pharmacokinetics of oral heroin administration demonstrated that the free drug and 6-acetylmorphine could not be detected in the arterial blood following dosing (Girardin et al., 2003). However, appreciable levels of morphine were detected, suggesting that an esterase present within the alimentary canal mediated the hydrolysis of heroin to this metabolite. Because high levels of hiCE (CES2) are present within the epithelial lining of the human gut, this suggests that this enzyme may be responsible for metabolism of heroin when this drug is taken orally. Our results, presented here, confirm that this enzyme is proficient in hydrolysing this drug, suggesting that in previous studies (Girardin et al., 2003), hiCE (CES2) present in the gut metabolized heroin immediately after absorption. This would account for the lack of the parent molecule present within the circulation.
As hiCE (CES2) has been identified as being proficient in the hydrolysis of heroin and the activation of CPT-11, potentially, selective CE inhibitors that prevent these hydrolytic reactions may prove useful in ameliorating the toxicity observed with these drugs. Therefore, we assessed the ability of a panel of benzil-based compounds and bis benzene-sulfonamides to modulate drug hydrolysis. As we have previously reported that these types of inhibitors act in a partially competitive fashion (Wadkins et al., 2004; Wadkins et al., 2005), the potency of enzyme inhibition will be dependent upon the substrate used for the analyses. Our results indicate however, that the phenylethane-1,2-dione based compounds that were chosen for analysis were potent inhibitors of all mammalian CEs, with all of the substrates tested (o-NPA, cocaine, heroin and CPT-11), whereas the sulfonamides only demonstrated selectivity for hiCE (CES2). The Ki values for enzyme inhibition were typically in the nM range, although the benzil analogues demonstrated greatest potency towards hiCE (CES2) when using CPT-11 as a substrate. We believe that selective administration of CE inhibitors after CPT-11 dosing, may alleviate the delayed diarrhea that results from intestinal drug activation. Because this toxicity is delayed up to 96 h following chemotherapy, this may provide a suitable window in which to inhibit hiCE (CES2). We are currently evaluating this hypothesis in animal models.
The studies described here demonstrate that hiCE (CES2) is efficient in hydrolysing cocaine and that inhibition of this enzyme by benzil-based compounds can be achieved in the presence of a variety of different clinically relevant substrates. These results should allow the development of approaches that may have utility in modulating the toxicity for such agents. For example, i.v. administration of hiCE (CES2) to rapidly convert cocaine to its inactive metabolites may be possible. Alternatively, selective hiCE (CES2) inhibitors may reduce drug-induced toxicity associated with CPT-11 hydrolysis in vivo.
Acknowledgments
We thank Dr J.P. McGovren for the kind gift of CPT-11. This work was supported in part by NIH Grants CA76202, CA79763, CA98468, CA108775, DA18116, a Cancer Center Core Grant CA21765 and by the American Lebanese Syrian Associated Charities.
Glossary
Abbreviations:
- BChE
butyrylcholinesterase
- CE
carboxylesterase
- clogP
calculated logP
- CPT-11, irinotecan
7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin
- DMSO
dimethyl sulfoxide
- hBChE
human BChE
- hBr3
human brain CE, CES3
- hCE1 (CES1)
human carboxylesterase 1, CES1
- hiCE (CES2)
human intestinal carboxylesterase, CES2, hCE2
- i
fractional inhibition
- IEC
ion-exchange chromatography
- kcat
turnover constant
- Ki
inhibition constant
- Km
Michaelis constant
- o-NPA
o-nitrophenyl acetate
- TBAP
tetrabutyl ammonium phosphate
- Vmax
maximal reaction velocity
Conflicts of interest
None.
References
- Anderson RN. Deaths: leading causes for 1999. Natl Vital Stat Rep. 1999;49:1–88. [PubMed] [Google Scholar]
- Beaufay H, Amar-Costesec A, Feytmans E, Thines-Sempoux D, Wibo M, Robbi M, et al. Analytical study of microsomes and isolated subcellular membranes from rat liver. I. Biochemical methods. J Cell Biol. 1974;61:188–200. doi: 10.1083/jcb.61.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM, et al. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer's drug tacrine. From binding promiscuity to selective inhibition. Chem & Biol. 2003a;10:341–349. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat Struct Biol. 2003b;10:349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- Bencharit S, Edwards CC, Morton CL, Howard-Williams EL, Kuhn P, Potter PM, et al. Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J Mol Biol. 2006;363:201–214. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski MR, Spink BJ, Dean RA, Berkman CE, Cashman JR, Bosron WF. Human liver carboxylesterase hCE-1: binding specificity for cocaine, heroin, and their metabolites and analogs. Drug Metab Dispos. 1997;25:1089–1096. [PubMed] [Google Scholar]
- Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- Danks MK, Morton CL, Pawlik CA, Potter PM. Overexpression of a rabbit liver carboxylesterase sensitizes human tumor cells to CPT-11. Cancer Res. 1998;58:20–22. [PubMed] [Google Scholar]
- Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, et al. Comparison of activation of CPT-11 by rabbit and human carboxylesterases for use in enzyme/prodrug therapy. Clin Cancer Res. 1999;5:917–924. [PubMed] [Google Scholar]
- Dodds HM, Rivory LP. The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11) Mol Pharmacol. 1999;56:1346–1353. doi: 10.1124/mol.56.6.1346. [DOI] [PubMed] [Google Scholar]
- Fleming CF, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, et al. Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J Mol Biol. 2005;352:165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Gatley SJ. Rapid stereoselective hydrolysis of (+)-cocaine in baboon plasma prevents its uptake in the brain: implications for behavioral studies. J Neurochem. 1990;54:720–723. doi: 10.1111/j.1471-4159.1990.tb01933.x. [DOI] [PubMed] [Google Scholar]
- Girardin F, Rentsch KM, Schwab MA, Maggiorini M, Pauli-Magnus C, Kullak-Ublick GA, et al. Pharmacokinetics of high doses of intramuscular and oral heroin in narcotic addicts. Clin Pharmacol Ther. 2003;74:341–352. doi: 10.1016/S0009-9236(03)00199-1. [DOI] [PubMed] [Google Scholar]
- Hicks LD, Hyatt JL, Stoddard S, Tsurkan L, Edwards CC, Wadkins RM, et al. Improved, selective, human intestinal carboxylesterase inhibitors designed to modulate 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (Irinotecan; CPT-11) toxicity. J Med Chem. 2009;52:3742–3752. doi: 10.1021/jm9001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humerickhouse R, Lohrbach K, Li L, Bosron W, Dolan M. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60:1189–1192. [PubMed] [Google Scholar]
- Hyatt JL, Stacy V, Wadkins RM, Yoon KJ, Wierdl M, Edwards CC, et al. Inhibition of carboxylesterases by benzil (diphenylethane-1,2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J Med Chem. 2005;48:5543–5550. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, et al. Planarity and constraint of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1. J Med Chem. 2007;50:5727–5734. doi: 10.1021/jm0706867. [DOI] [PubMed] [Google Scholar]
- Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J Pharmacol Expt Ther. 1996;279:713–717. [PubMed] [Google Scholar]
- Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of CPT-11 by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725–4728. [PubMed] [Google Scholar]
- Lockridge O, LaDu BN. Diacetylmorphine (heroin) hydrolysis by human-serum cholinesterase (pseudocholinesterase or butyrylcholinesterase) Fed Proc. 1980;39:1754. [Google Scholar]
- Mori M, Hosokawa M, Ogasawara Y, Tsukada E, Chiba K. cDNA cloning, characterization and stable expression of novel human brain carboxylesterase. FEBS Lett. 1999;458:17–22. doi: 10.1016/s0014-5793(99)01111-4. [DOI] [PubMed] [Google Scholar]
- Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda and COS7 cells for recombinant gene expression: application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- Morton CL, Wadkins RM, Danks MK, Potter PM. CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase. Cancer Res. 1999;59:1458–1463. [PubMed] [Google Scholar]
- Munger JS, Shi GP, Mark EA, Chin DT, Gerard C, Chapman HA. A serine esterase released by human alveolar macrophages is closely related to liver microsomal carboxylesterases. J Biol Chem. 1991;266:18832–18838. [PubMed] [Google Scholar]
- Pindel EV, Kedishvili NY, Abraham TL, Brzezinski MR, Zhang J, Dean RA, et al. Purification and cloning of a broad substrate specificity human liver carboxylesterase that catalyzes the hydrolysis of cocaine and heroin. J Biol Chem. 1997;272:14769–14775. doi: 10.1074/jbc.272.23.14769. [DOI] [PubMed] [Google Scholar]
- Potter PM, Wadkins RM. Carboxylesterases – detoxifying enzymes and targets for drug therapy. Curr Med Chem. 2006;13:1045–1054. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- Potter PM, Wolverton JS, Morton CL, Wierdl M, Danks MK. Cellular localization domains of a rabbit and a human carboxylesterase: influence on irinotecan (CPT-11) metabolism by the rabbit enzyme. Cancer Res. 1998;58:3627–3632. [PubMed] [Google Scholar]
- Redinbo MR, Potter PM. Mammalian carboxylesterases: from drug targets to protein therapeutics. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TJ. Comparitive protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schwer H, Langmann T, Daig R, Becker A, Aslanidis C, Schmitz G. Molecular cloning and characterization of a novel putative carboxylesterase, present in human intestine and liver. Biochem Biophys Res Comm. 1997;233:117–120. doi: 10.1006/bbrc.1997.6413. [DOI] [PubMed] [Google Scholar]
- Stewart DJ, Inaba T, Tang BK, Kalow W. Hydrolysis of cocaine in human plasma by cholinesterase. Life Sci. 1977;20:1557–1563. doi: 10.1016/0024-3205(77)90448-9. [DOI] [PubMed] [Google Scholar]
- Tabata T, Katoh M, Tokudome S, Nakajima M, Yokoi T. Identification of the cytosolic carboxylesterase catalyzing the 5′-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab Dispos. 2004;32:1103–1110. doi: 10.1124/dmd.104.000554. [DOI] [PubMed] [Google Scholar]
- Wadkins RM, Morton CL, Weeks JK, Oliver L, Wierdl M, Danks MK, et al. Structural constraints affect the metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol Pharmacol. 2001;60:355–362. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]
- Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, et al. Identification of novel selective human intestinal carboxylesterase inhibitors for the amelioration of irinotecan-induced diarrhea: synthesis, quantitative structure-activity relationship analysis, and biological activity. Mol Pharmacol. 2004;65:1336–1343. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, et al. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J Med Chem. 2005;48:2905–2915. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- Webb JL. Enzyme and Metabolic Inhibitors. Volume 1. General Principles of Inhibition. New York: Academic Press Inc; 1963. [Google Scholar]
- Wierdl M, Tsurkan L, Hyatt JL, Hatfield MJ, Edwards CC, Danks MK, et al. An improved human carboxylesterase for use in enzyme/prodrug therapy with CPT-11. Cancer Gene Ther. 2008;15:183–192. doi: 10.1038/sj.cgt.7701112. [DOI] [PubMed] [Google Scholar]
- Wu MH, Chen P, Remo BF, Cook EH, Jr, Das S, Dolan ME. Characterization of multiple promoters in the human carboxylesterase 2 gene. Pharmacogenetics. 2003;13:425–435. doi: 10.1097/00008571-200307000-00008. [DOI] [PubMed] [Google Scholar]
- Zhang J, Burnell JC, Dumaual N, Bosron WF. Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J Pharmacol Exp Ther. 1999;290:314–318. [PubMed] [Google Scholar]
- Zhu HJ, Patrick KS, Yuan HJ, Wang JS, Donovan JL, DeVane CL, et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am J Hum Genet. 2008;82:1241–1248. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]