

Abstract
BACKGROUND AND PURPOSE
Acute exacerbations of chronic obstructive pulmonary disease (COPD), which are often associated with respiratory infections, are defined as a worsening of symptoms that require a change in medication. Exacerbations are characterized by a reduction in lung function, quality of life and are associated with increased pro-inflammatory mediators in the lung. Our aim was to develop an animal model to mimic aspects of this exaggerated inflammatory response by combining key etiological factors, tobacco smoke (TS) and bacterial lipopolysaccharide (LPS).
EXPERIMENTAL APPROACH
Rats were exposed to TS for 30 min twice a day for 2 days. On day 3 animals were exposed to LPS for 30 min followed by exposure to TS 5 h later. Inflammation, mucus and lung function were assessed 24 h after LPS.
KEY RESULTS
Neutrophils, mucus, oedema and cytotoxicity in lung and/or bronchoalveolar lavage was increased in animals exposed to combined LPS and TS, compared with either stimulus alone. Lung function was impaired in animals exposed to combined LPS and TS. Inflammatory cells, oedema and mucus were unaffected by pretreatment with the corticosteroid, budesonide, but were reduced by the phosphodiesterase 4 selective inhibitor roflumilast. Additionally, lung function was improved by roflumilast.
CONCLUSIONS AND IMPLICATIONS
We have established an in vivo model mimicking characteristic features of acute exacerbations of COPD including lung function decline and increased lung inflammation. This model may be useful to investigate molecular and cellular mechanisms underlying such exacerbations, to identify new targets and to discover novel therapeutic agents.
Keywords: acute exacerbations; COPD; tobacco smoke; LPS, steroids; PDE4 inhibitors
Introduction
An acute exacerbation of chronic obstructive pulmonary disease (COPD) is defined as ‘an event in the natural course of the disease that is characterized by a change in the patient's baseline dyspnea, cough or sputum beyond day-to-day variability and sufficient to warrant a change in management’ (Pauwels et al., 2001; Celli and MacNee, 2004). An increased frequency of COPD exacerbations is associated with a reduction in patient health status resulting in a major detrimental effect on acute morbidity and mortality (Seemungal et al., 1998). Current therapeutic agents have limited effects and thus acute exacerbations requiring hospitalization are a significant burden on healthcare services (Sullivan et al., 2000; Calverley, 2008). Discovering effective therapeutic interventions for acute exacerbations of COPD should improve patient quality of life and also reduce the disease burden.
The most common causes of an acute exacerbation are thought to be viral and bacterial infections. Indeed a study recently demonstrated that respiratory viral and/or bacterial infections were isolated from 78% of COPD patients admitted to hospital with exacerbations of their disease (Papi et al., 2006). Specifically, the Gram negative bacteria, non-typeable Haemophilus influenzae and Moraxella catarrhalis, are two of the most commonly isolated bacterial pathogens from patients during exacerbations (Sethi and Murphy, 2001).
During an exacerbation, COPD patients have elevated levels of inflammatory mediators in sputum compared with patients with stable COPD. Studies have shown an increase in inflammatory cells including neutrophils, eosinophils and T-lymphocytes in sputum, BAL and/or lung tissue (Balbi et al., 1997; Drost et al., 2005; Fujimoto et al., 2005; Tsoumakidou et al., 2005). These increases in inflammatory cells are associated with an increase in mucus and other inflammatory mediators and cytokines(Aaron et al., 2001; Gompertz et al., 2001; Fujimoto et al., 2005). As well as a transient increase in inflammation during exacerbations, the exacerbations appear to result in a small but significant increase in the rate of lung function decline compared with non-exacerbating COPD patients (Donaldson et al., 2002).
As in stable COPD, this exaggerated inflammatory response during an acute exacerbation is treated with corticosteroids. Clinical trials suggest that oral corticosteroids may have beneficial effects reducing exacerbation rate and the associated morbidity (Davies et al., 1999; Niewoehner et al., 1999). Inhaled corticosteroids have also been shown to reduce the frequency of exacerbations in COPD patients (Pauwels et al., 1999; Vestbo et al., 1999; Burge et al., 2000; Alsaeedi et al., 2002). The inflammation observed in COPD patients is well recognized to be less responsive to steroids than that seen in asthma (Adcock and Barnes, 2008); indeed the levels of neutrophils and cytokines in the sputum have been shown to be unaffected by high doses of inhaled corticosteroids in COPD patients (Culpitt et al., 1999). In addition to corticosteroids, the effect of the selective phosphodiesterase (PDE)4 inhibitor, roflumilast, has been evaluated in the clinic in COPD patients (Spina, 2008). First, in stable COPD patients, roflumilast had some anti-inflammatory effects and caused a small improvement in lung function (Rabe et al., 2005; Grootendorst et al., 2007). Interestingly, in a study in which patients with severe COPD were treated with roflumilast, for 1-year, a decreased exacerbation rate in the most severe COPD patients (GOLD stage IV) was observed. However, when the whole cohort was included, there was no significant reduction in exacerbation rate (Calverley et al., 2007). As exacerbations are thought to play an important role in the progression of COPD, the identification of novel therapies which have more pronounced benefits in the treatment of exacerbations would be expected to lead to substantial improvements in the patient's quality of life and mortality.
Little is known about the cellular and molecular mechanisms underlying acute exacerbations of COPD and thus there is a requirement to develop suitable model systems to provide insights and facilitate the discovery of novel therapeutic agents. Studies have been performed in mice by exposing them to a combination of tobacco smoke (TS) and bacterial lipopolysaccharide (LPS). These studies demonstrate that mice exhibit a different inflammatory profile when exposed to LPS in combination with TS compared with animals exposed to either LPS or smoke alone (Meng et al., 2006; Lee et al., 2007). For example an increase in BAL neutrophils together with an increase in lung tissue apoptosis was observed in mice exposed to the combination of LPS and TS versus either stimulus alone (Lee et al., 2007).
The objective of our study was to establish an acute model system to try and reproduce aspects of the increased inflammatory response seen during an acute exacerbation of COPD by exposing Sprague-Dawley rats to a combination of LPS and TS. In addition to inflammatory cells, we measured mucus levels and various lung function parameters. To further characterize this model, we tested the effects of two known anti-inflammatory agents, a corticosteroid, budesonide and the phosphodiesterase (PDE)4 inhibitor roflumilast. Enhanced inflammation and reduced lung function was observed in the rats exposed to a combination of LPS and TS versus rats exposed to either stimulus alone. In addition, the inflammation induced by a combination of LPS and TS was insensitive to steroids, whereas a PDE4 inhibitor was effective at reducing the inflammation and improving lung function parameters. It is anticipated that this model will provide insights into the pathways involved in the enhanced inflammatory response observed during an acute exacerbation of COPD, and thus facilitate the identification of novel effective therapeutic agents.
Methods
In vivo protocols
All animal care and experimental studies were performed under a Project License issued by the United Kingdom Home Office under the Animal (Scientific Procedures) Act 1986 and approved by local ethical review processes (ERP). Adult male Sprague-Dawley rats (250–350 g; eight per treatment group) were housed in rooms maintained at constant temperature (21 ± 2°C) and humidity (55 ± 15%) with a 12 h light cycle and 15–20 air changes per hour. Animals were allowed food, RM1 Pellets (SDS UK Ltd) and water ad libitum.
In these studies inflammation was induced by the combination of LPS and TS, LPS alone or TS alone. For these studies rats were exposed to TS (1R3F Kentucky Research cigarettes) for 30 min twice a day on two consecutive days with at least a 5 h gap between each exposure. On the morning of day 3 animals were exposed for 30 min to aerosolized LPS (E. coli serotype 0111:B4; 0.3 mg·mL−1) generated by a Topaz ATM210 compressed air aerosol generator. Five hours after LPS exposure, animals were exposed to TS for 30 min. In the groups in which rats were exposed to TS alone, the same protocol was followed, but instead of aerosolized LPS, rats were exposed to a saline aerosol generated in the same manner for 30 min. In the groups exposed to LPS alone the same protocol was followed with rats exposed to room air for 30 min in the place of each TS exposure. Budesonide (prepared in 0.5% NaCMC), roflumilast [prepared in 70% PEG300/30% Glucose (5% w/v)] or the respective vehicles were given orally (0.5 mL per rat) 1 hour prior to each exposure to either air/TS or saline/LPS. In the satellite study where lung function was assessed, rats were treated orally (0.5 mL per rat) 1 hour prior to each smoke exposure on day 1 and then 1 hour prior to the first smoke exposure on days 2 and 3. The doses and route of administration of budesonide and roflumilast were selected based on what has been reported in other in vivo model systems (Birrell et al., 2005; Wollin et al., 2006).Animals were killed with an overdose of euthatal (200 mg i.p) 24 h after the saline/LPS exposure.
Bronchoalveolar lavage fluid and lung tissue analysis
Bronchoalveolar lavage (BAL) was performed by instilling 3 × 4 mL of sterile phosphate buffered saline (PBS) (without calcium and magnesium) into the airways through a cannula and then recovering the PBS. Cytospins were prepared for differential cell count analysis by centrifuging aliquots of BAL fluid in a cytospin at 55×g with low acceleration at room temperature for 5 min. The slides were then fixed and stained on a Bayer Hema-Tek using modified Wright Giemsa stain. The remaining BAL fluid was spun at 300 g for 10 min at 4°C. The supernatant was removed and stored at −80°C until required for analysis of total protein and lactate dehydrogenase activity. The cell pellet was re-suspended in methyl-violet for analysis of total cell counts using a haemocytometer. Differential cell counts of the cells recovered from the airway lumen were determined by light microscopy from the cytospin preparations using standard morphological criteria and the percentage of macrophages, lymphocytes and neutrophils were determined. The concentration of the total protein in the BAL was determined using a bicinchoninic acid (BCA) protein assay kit following manufacturer's instructions (Thermoscientific, Cramlington, UK). Lactate dehydrogenase activity was assessed using a Cytotoxicity detection kit following manufacturer's instructions (Roche Diagnostics, Burgess Hill, UK).
Immediately after BAL, the top right lung lobe was removed and enzymically digested using a method modified from the literature (Underwood et al., 1997). The lung lobe was perfused with room temperature RPMI 1640 using a peristaltic pump to remove the blood. The tissue was chopped finely using a McIllwain chopper and incubated for 1 h at 37°C with gentle agitation in 10 mL RPMI 1640/10% FBS containing collagenase (1 mg·mL−1) and DNase (25 µg·mL−1) in a shaking water bath. The recovered cells were filtered through a cell sieve (mesh size 70 µm) and washed three times with room temperature RPMI 1640. Total and differential cell counts were performed on the cells that were recovered. The remaining right lung lobes were tied off and snap frozen in liquid nitrogen. Total RNA was isolated from lung tissue using the RNeasy mini RNA isolation kit (Qiagen™). First strand cDNA was prepared using the first strand cDNA synthesis kit (Applied Biosystems). For TaqMan reverse transcriptase-PCR, inventoried TaqMan gene expression assays for β-actin and MUC5AC were purchased from Applied Biosystems and experiments performed using an ABI PRISM 7900 sequence detector.
Histology
The left lung lobe was insufflated with 10% neutral buffered formalin (NBF) at a constant pressure of 25 cm H2O for histological assessment. Lung tissue neutrophils were identified by staining 3 µm lung sections using substituted Napthol AS-D chloroacetate esterase method in paraffin sections. Changes in mucus levels were detected using fluorescein conjugated Ulex europaeus agglutinin-1 (UEA-1) (Vector Laboratories, Peterborough, UK) in conjunction with a secondary antibody, Biotin-SP IgG fraction monoclonal mouse anti-FITC (Jacksons Immunoresearch, Newmarket, UK). Immunohistochemistry was performed on an automated immunostainer (Ventana Medical Systems, Tuscon, AZ, USA) using heat induced antigen retrieval and standard DAB detection kit (Ventana Medical Systems). The area of UEA-1 staining was analysed on between 10 to 15 airway fields for each sample with a KS400 image analyser (Image Associates, Congleton, UK). UEA-1 has been previously validated as a marker of respiratory mucus levels (Jackson et al. 2002).
Lung function assessment
Lung function was measured using a forced manoeuvres system (Buxco, Winchester, UK). Rats were anaesthetized (medetomidine 0.33 mg·kg−1·ketamine−1 50 mg·kg−1, 1 mL·kg−1, i.p.) and the trachea cannulated. The tracheal cannula was connected up to a manifold and a volume history manoeuvre was performed three times. This ensures all rats have a similar pretreatment regarding pulmonary inflation pressure. Following this, three fast flow manoeuvres were performed with at least a one-minute interval between them. This manoeuvre involved inflating the animal's lungs to a tracheal pressure of 25 cmH2O, holding this pressure for 2 s and then exhaling as quickly as possible, until the respiratory flow declined to 5 mL·s−1. Total lung capacity (TLC), forced vital capacity (FVC) and forced expiratory volume in the first 100 ms (FEV100) were calculated from this manoeuvre. Subsequently, a quasistatic pressure volume manoeuvre was performed with the rat inspired to TLC and then slowly expired to residual volume. The slope of the expiration curve between 0–10 cm H2O was measured (chord compliance; Cchord).
Statistics
All of the data are expressed as mean ± standard error of the mean (SEM). The statistical analysis performed in these studies were those suggested by a qualified statistician and based on hypotheses generated prior to the initiation of the studies. In Figures 1–4 comparisons are made between each group, but only the statistically significant differences are shown. In cases where two groups were compared a Mann-Whitney U-test was performed. Multiple group comparisons were performed using the Kruskal-Wallis test followed by the Dunn's multiple comparison post test. P < 0.05 was taken to indicate statistical significance.
Figure 1.
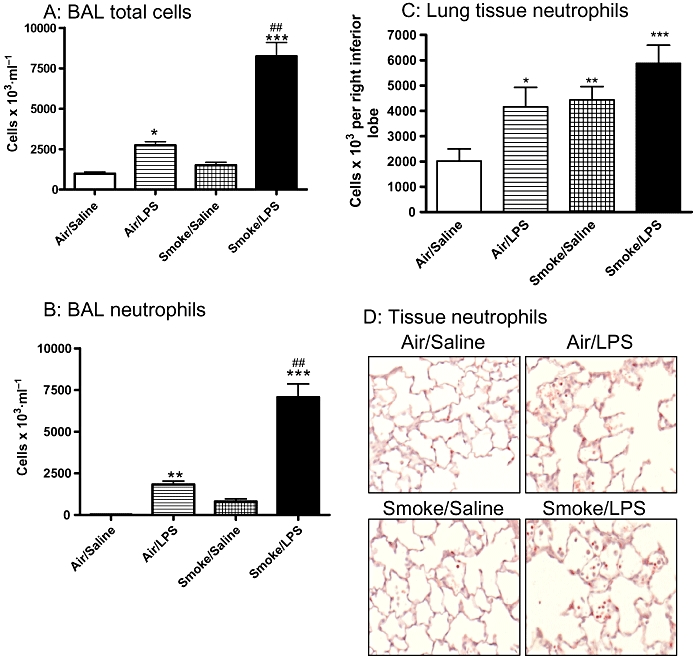
Inflammatory cell numbers are elevated in response to exposure to a combination of LPS and tobacco smoke (TS). Sprague-Dawley rats were exposed to TS or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by TS or air 5 h later. Twenty-four hours after LPS exposure the number of (A) total cells were assessed in the BAL and the numbers of neutrophils were assessed in the (B) BAL and lung tissue using (C) tissue digests. Data are expressed as the mean and SEM of n= 8 animals per group. (D) Neutrophils were also assessed in the lung tissue by histological assessment. Representative sections from each group (8 animals per group) are shown. (*P < 0.05, **P < 0.01, ***P < 0.001 compared with air/saline controls; ##P < 0.01 compared with TS/saline group). BAL, broncho alveolar lavage; LPS, lipopolysaccharide.
Materials
Sprague-Dawley rats (Charles-River, Margate, UK), RM1 pellets (SDS UK Ltd., Witham, UK), 1R3F Research cigarettes (Kentucky University, Lexington, KT, USA), budesonide, modified Wright-Giemsa stain and LPS (E. coli 0111:B4) (Sigma, Dorset, UK), roflumilast (Tocris, Bristol, UK), PBS, RPMI 1640, fetal bovine serum (FBS) (Invitrogen, Paisley, UK), BCA protein assay kit (Thermoscientific), cytotoxicity detection kit, collagenase, DNase (Roche Diagnostics), RNeasy mini RNA isolation kit (Qiagen, Crawley, UK), cDNA synthesis kit, gene expression assays (Applied Biosystems, Warrington, UK).
Results
Effect of LPS and TS challenge on inflammation in Sprague-Dawley rats
Sprague-Dawley rats challenged with a combination of LPS and TS showed a marked increase in total inflammatory cells in the airway lumen compared with animals challenged with either inflammatory stimulus individually (TS/LPS: 8240 ± 854 × 103 cells·mL−1 vs. air/LPS: 2737 ± 223 × 103 cells·mL−1, TS/saline: 1496 ± 189 × 103 cells·mL−1 or air/saline: 977 ± 109 × 103 cells·mL−1). The increase in total cells comprised mainly of an increase in neutrophil numbers, which were significantly elevated after exposure to the combination of LPS and TS (7070 ± 760 cells × 103·mL−1) compared with TS alone (807 ± 157 cells × 103·mL−1 for the TS/LPS (Figure 1A and B). In the lung tissue the levels of neutrophils determined by histological assessment and tissue digests were highest in animals exposed to a combination of LPS and TS; however, there was not a statistically significant difference compared with those exposed to either stimulus alone (Figure 1C and D).
Effect of LPS and TS on lung mucus levels in Sprague-Dawley rats
In addition to an increase in inflammatory cells the levels of mucus in the airways were increased after challenge with a combination of LPS and TS compared with either LPS or TS stimulus alone; however, this difference was not statistically significant (Figure 2A and B). The increase in the level of mucus was associated with an increase in MUC5AC mRNA levels, compared with air/saline controls (Figure 2C).
Figure 2.
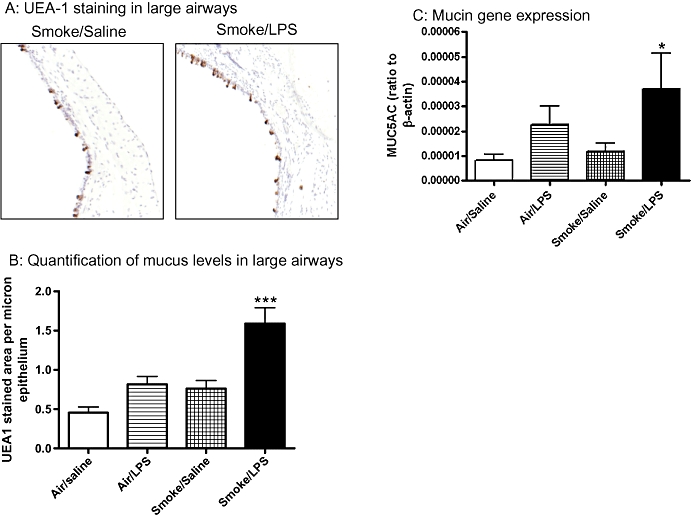
Mucus levels are elevated in response to exposure to a combination of LPS and tobacco smoke. Sprague-Dawley rats were exposed to tobacco smoke or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by tobacco smoke or air 5 h later. Twenty-four hours after LPS exposure the levels of mucus were determined by UEA-1 staining and MUC5AC gene expression. Data shown are (A) representative pictures of UEA-1 staining (B) quantification of UEA-1 stained area expressed as mean and SEM of n= 8 per group and (C) MUC5AC gene expression levels in the lung tissue expressed as mean and SEM of n= 7–8 per group. (*P < 0.05, ***P < 0.001 compared with air/saline controls). LPS, lipopolysaccharide; MUC5AC, muucin-5AC; UEA-1, Ulex europaeus agglutinin-1.
Effect of LPS and TS challenge on cytotoxicity and plasma protein extravasation in the lungs of Sprague-Dawley rats
Lactate dehydrogenase (LDH) activity was significantly elevated in the BAL fluid of animals exposed to LPS and TS versus those exposed to LPS alone, suggesting that the combination of LPS and TS causes an increase in cytotoxicity (Figure 3A). The levels of total protein detected in the BAL fluid were also significantly enhanced in animals challenged with a combination of LPS and TS compared with those challenged with LPS alone, suggesting that there is increase in plasma protein extravasation (Figure 3B).
Figure 3.
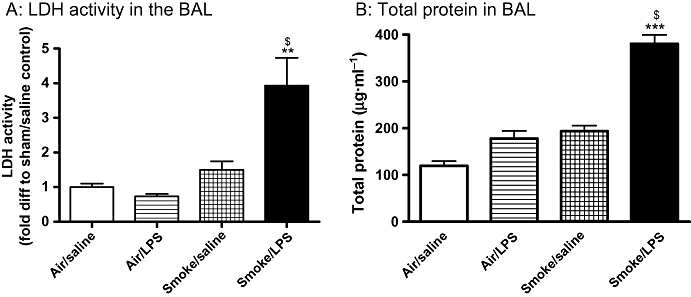
Lipopolysaccharide (LPS) and tobacco smoke (TS) exposure results in an increase in cytotoxicity and oedema. Sprague-Dawley rats were exposed to TS or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by TS or air 5 h later. Twenty-four hours after LPS exposure the levels of (A) LDH activity and (B) total protein in the BAL were assessed. Data are expressed as mean and SEM of n= 8 per group. **P < 0.01, ***P < 0.001 compared with air/saline controls; $P < 0.05 compared with air/LPS comtrols). BAL, broncho alveolar lavage.
Effect of LPS and TS challenge on lung function in Sprague-Dawley rats
The effect of challenging animals with a combination of LPS and TS on lung function was also investigated. No change was observed in lung function parameters in the animals exposed to TS alone. There was an apparent drop in lung function in rats exposed to LPS alone; however, this did not reach statistical significance. In contrast, challenge with a combination of LPS and TS led to a significant reduction in total lung capacity (TLC), chord compliance and forced expiratory volume in 100 ms (FEV100) compared with air/saline challenged animals (Figure 4A–C respectively).
Figure 4.
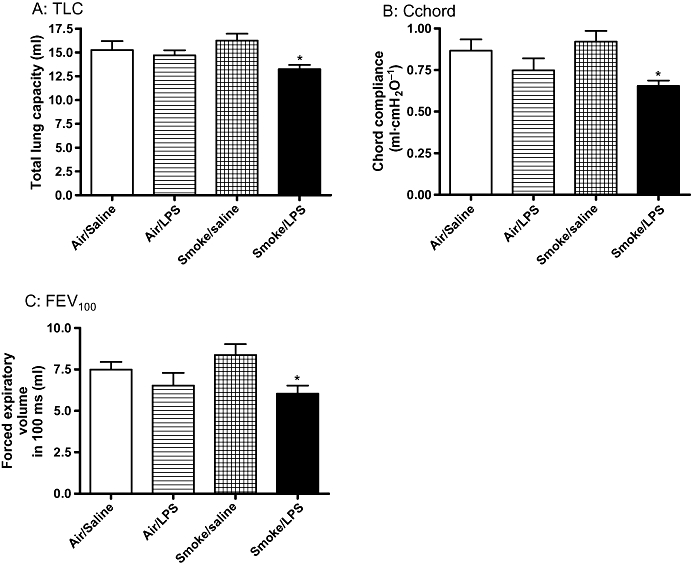
Lung function parameters are impaired in LPS and TS exposed rats. Sprague-Dawley rats were exposed to tobacco smoke or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by tobacco smoke or air 5 h later. Twenty-four hours after LPS exposure (A) total lung capacity (TLC) (B) chord compliance (Cchord) and (C) FEV100 were assessed by forced manoeuvres. Data are expressed as mean and SEM of n= 8 animals per group. (*P < 0.05 compared with air/saline exposed animals). LPS, lipopolysaccharide; TS, tobacco smoke.
Effect of budesonide on LPS- and TS-induced inflammation, mucus and protein levels
To determine whether the inflammation in response to exposure to LPS and TS was responsive to corticosteroids we tested the effects of orally administered budesonide. Budesonide (3 mg·kg−1) significantly reduced the numbers of neutrophils in the BAL fluid recovered from LPS-challenged animals (Figure 5A). In animals exposed to smoke alone, budesonide (3 mg·kg−1) did not significantly affect the number of neutrophils in the BAL fluid. Budesonide also did not significantly affect the increase in BAL neutrophils in animals exposed to a combination of LPS and TS (Figure 5A). Furthermore, budesonide did not significantly alter mucus or total protein levels following exposure to LPS and TS (Figure 5B and C)
Figure 5.
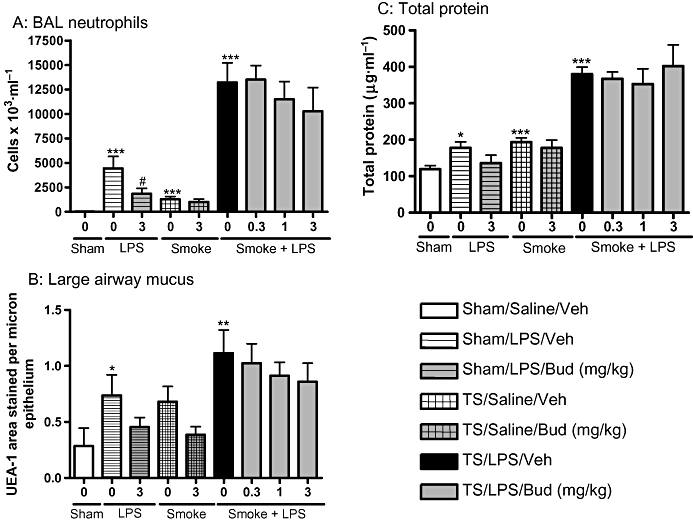
Budesonide does not significantly affect inflammation induced by the combination of LPS and TS. Sprague-Dawley rats were exposed to tobacco smoke or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by tobacco smoke or air 5 h later. One hour prior to each exposure animals were either dosed with vehicle or budesonide (0.3, 1 or 3 mg·kg−1). Twenty-four hours after LPS exposure (A) neutrophils (B) mucus and (C) total protein levels were assessed. Data represent the mean and SEM of n= 8 animals per group which was performed in one single experiment. (*P < 0.05, **P < 0.01, ***P < 0.001 compared with air/saline controls; #P < 0.05 compared with appropriate stimulus/vehicle control). BAL, broncho alveolar lavage; LPS, lipopolysaccharide; TS, tobacco smoke; UEA-1, Ulex europaeus agglutinin-1.
Effect of roflumilast on LPS and TS induced inflammation, mucus and protein levels
The effects of the selective PDE4 inhibitor, roflumilast, on LPS and TS induced lung inflammation, mucus and protein levels were assessed. Roflumilast (10 mg·kg−1) significantly attenuated the increase in total cells in BAL induced by a combination of LPS and TS, compared with vehicle controls (5828 ± 650 × 103·mL−1 vs. 11 459 ± 1951 × 103·mL−1 respectively; P < 0.05) but had no statistically significant inhibitory effect on BAL neutrophil numbers (Figure 6A). Roflumilast (10 mg·kg−1), however, did cause a statistically significant reduction in mucus and total protein levels in animals exposed to a combination of LPS and TS (Figure 6B and C). The effects of roflumilast (10 mg·kg−1) on lung function parameters were assessed in a separate study. A significant improvement in TLC, FEV100, Cchord and forced vital capacity (FVC) were observed in animals treated with roflumilast at 10 mg·kg−1 (Figure 7).
Figure 6.
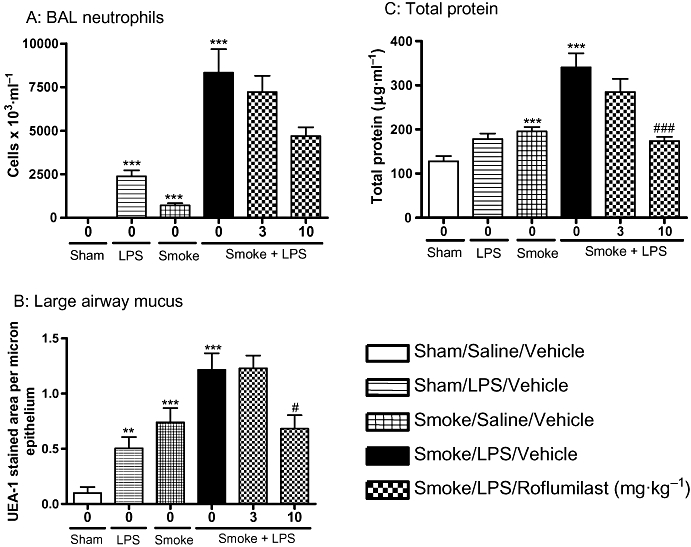
Roflumilast significantly reduced the inflammatory mediators induced by the combination of LPS and TS. Sprague-Dawley rats were exposed to tobacco smoke or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by tobacco smoke or air 5 h later. One hour prior to each exposure animals were either dosed with vehicle or roflumilast (3 or 10 mg·kg−1). Twenty-four hours after LPS exposure (A) neutrophils (B) mucus and (C) total protein levels were assessed. Data represent the mean and SEM of n= 8 animals per group which was performed in one single experiment. (**P < 0.01, ***P < 0.001 compared with air/saline controls; #P < 0.05, ###P < 0.001 compared with LPS/smoke/vehicle control). BAL, broncho alveolar lavage; LPS, lipopolysaccharide; TS, tobacco smoke; UEA-1, Ulex europaeus agglutinin-1.
Figure 7.
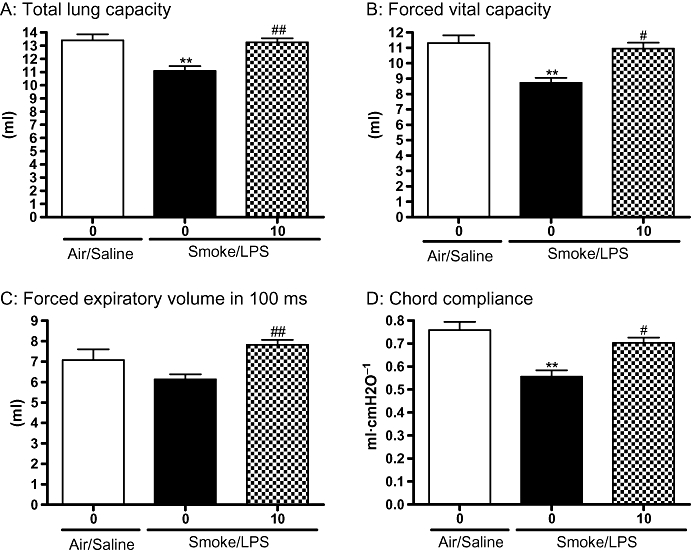
Roflumilast significantly improved lung function parameters impaired by the combination of lipopolysaccharide (LPS) and tobacco smoke (TS). Sprague-Dawley rats were exposed to TS or air for 30 min twice a day for 2 days. On the morning of day 3 rats were exposed to saline of LPS (0.3 mg·mL−1) for 30 min followed by TS or air 5 h later. Animals were dosed with roflumilast (10 mg·kg−1) twice on day one and then once daily on days 2 and 3. Twenty-four hours after LPS exposure (A) TLC (B) FVC (C) FEV100 and (D) chord compliance were assessed. Data represent the mean and SEM of n= 7–8 animals per group which was performed in one single experiment. (**P < 0.01 compared with air/saline/vehicle controls; #P < 0.05, ##P < 0.01 compared with LPS/smoke/vehicle control). FEV100, forced expiratory volume in the first 100 milliseconds; FVC, forced vital capacity; TLC, total lung capacity.
Discussion
Acute exacerbations of COPD are associated with an increase in patient morbidity and mortality. Currently there are no effective therapies for the treatment of acute exacerbation of COPD and the molecular mechanisms are poorly understood. It is therefore important to develop in vivo models to aid with the identification of novel therapeutic targets, and ultimately effective therapies. In this study we have developed a model system to try and reproduce aspects of an acute exacerbation of COPD by combining the main causative agent of COPD, TS, with LPS, to mimic aspects of a bacterial-induced acute exacerbation. We have demonstrated that exposing animals to a combination of LPS and TS results in increased inflammatory cells, mucus, cytotoxicity and total protein in the lung as well as a reduction in lung function compared with the air/saline control. In some cases the increase in inflammation was significantly elevated compared with either smoke or LPS exposure alone. The enhanced inflammation, mucus and total protein levels while insensitive to the corticosteroid budesonide, were reduced by the PDE4 inhibitor, roflumilast.
The inflammation present during the course of acute exacerbations has been assessed in COPD patients. During an acute exacerbation there is evidence that patients have elevated sputum neutrophils and mucus hypersecretion, which have both been associated with a decrease in FEV1 (Vestbo et al., 1996; Miravitlles et al., 2000; Drost et al., 2005; Miravitlles et al., 2006). Additionally during an acute exacerbation, significant decreases in lung function parameters are observed which are at least partially reversed upon recovery. Studies have demonstrated that the greater the extent of lung function decline, the longer it takes to recover from the exacerbation (Seemungal et al., 2000). To our knowledge, the cause of this reversible change in lung function during an exacerbation in COPD patients has not been elucidated.
In this model we have demonstrated that exposing Sprague-Dawley rats to a combination of LPS and TS results in an increase in lung neutrophil numbers, a decrease in lung function parameters and an increase in airway mucus levels. An earlier study had demonstrated that exposing animals to a combination of LPS and TS resulted in a change in the inflammatory response compared with exposure to either stimulus alone (Meng et al., 2006; Lee et al., 2007). Similar to the findings in our study, the group demonstrated that combined LPS and TS exposure produces an exaggerated inflammatory response (and increased cytotoxicity) compared with animals exposed to either stimuli alone (Lee et al., 2007). Additionally a recent paper has demonstrated that exposing mice to TS for 8 weeks followed by innoculation with non-typeable Haemophilus Influenza (NTHI) resulted in a similar greater than additive increase in neutrophils in the BAL compared with either stimulus alone (Gaschler et al., 2009).
In this study as well as observing an increase in inflammatory cells we have extended these findings to demonstrate that total lung capacity, FEV100 and chord compliance were reduced in rats exposed to LPS and TS compared with air/saline exposed controls. Interestingly in this study we were able to detect an increase in the levels of total protein in the BAL fluid in animals exposed to a combination of LPS and TS compared with those exposed to either stimulus alone. As Baginski et al. (2006), we also observed a greater than additive increase in mucus in rats exposed to the combination of LPS and TS compared with those exposed to either stimulus alone. We therefore hypothesize that the reduction in lung function in our model may be due to an increase in plasma protein extravasation leading to oedema and/or mucus hypersecretion.
To further characterize this model we tested the effects of known anti-inflammatory compounds, corticosteroids and PDE4 inhibitors, which have been investigated in COPD patients. Corticosteroids have been show to reduce the duration and frequency of exacerbations (Davies et al., 1999; Niewoehner et al., 1999; Pauwels et al., 1999; Vestbo et al., 1999; Burge et al., 2000); but have limited effects on sputum neutrophils (Culpitt et al., 1999; Davies et al., 1999; Niewoehner et al., 1999). PDE4 inhibitors show modest, but significant improvements in quality of life scores and exacerbation frequency in COPD patients (Rabe et al., 2005; Calverley et al., 2007; Grootendorst et al., 2007). Additionally a reduction in the levels of sputum neutrophils and CD8 + ve T cells has been observed in COPD patients after treatment with roflumilast (Grootendorst et al., 2007).
In these studies, budesonide (3 mg·kg−1) significantly reduced the numbers of neutrophils in the BAL fluid in the rats exposed to LPS alone demonstrating that this dosing regimen facilitated sufficient exposure to induce anti-inflammatory effects. In contrast, at the same dose of budesonide (3 mg·kg−1), the inflammation induced by TS alone was not significantly attenuated. Our results are similar to those obtained by, Leclerc et al. (2006) who demonstrated that while dexamethasone inhibited LPS-induced inflammation in mice, it failed to reduce the increase in neutrophils induced by TS. In addition, in our study, budesonide did not significantly affect the inflammation induced by the combination of LPS and TS.In contrast to this finding, Gaschler et al. (2009) demonstrated that dexamethasone attenuated the inflammation induced by TS and NTHI. The differences in corticosteroid sensitivity between these models are not fully understood; however, the protocols used differ in species, duration, type and route of administration of corticosteroid, and the fact that Gaschler et al. (2009) used live bacteria rather than LPS. All of these differences may alter corticosteroid sensitivity and higher doses of budesonide may be effective.
In the studies reported here the trend towards a reduction in neutrophil numbers and the statistically significant reduction in plasma protein extravasation and mucus levels provide evidence that roflumilast could inhibit the inflammatory response elicited by the combination of LPS and TS. It is possible that the lack of a statistically significant reduction in neutrophils is due to the variability of the data. While we did not evaluate the effect of roflumilast on the LPS or smoke induced inflammation alone, the PDE4 inhibitor, cilomilast, has been shown to be capable of suppressing inflammation induced by either LPS or TS alone (Leclerc et al., 2006). Additionally in these studies we demonstrate the reduction in lung function parameters induced by exposure to the combination of LPS and TS was significantly reversed by the PDE4 inhibitor roflumilast. These findings further support our hypothesis that the reduction in lung function parameters associated with exposure to the combination of LPS and smoke may be due to mucus hypersecretion and/or plasma protein extravasation. In the future it would be of value to determine the effect of these and other pharmacological agents on the duration of the increased inflammatory response.
In this study LPS, a component of the cell wall from Gram negative bacteria, was used to mimic the induction of inflammation by a bacterial infection through the Toll-like receptors, TLR4. However, during an exacerbation, in addition to signalling through TLR4, the bacterial infection will induce an inflammatory response through a number of different pathogen recognition receptors (PRRs). Clearly another caveat to this model is that the inflammatory response is induced in 3 days and therefore can not replicate the chronicity of the inflammation observed in COPD patients which develops over many years prior to an acute exacerbation. However, the exaggerated inflammatory response, decline in lung function and increased mucus observed in this model are similar to certain aspects of that observed in COPD patients during an acute exacerbation of their disease. As this model mimics aspects of the exaggerated inflammatory response observed during an acute exacerbation, it may be useful to elucidate the underlying molecular mechanisms that are involved. If these prove to be the same in COPD patients, then it may aid in the discovery of novel effective therapies for treatment of acute exacerbations of COPD.
Acknowledgments
We would like to thank all the members of Kathy Banners team and the histology team for their help with these studies. Additionally, we would like to thank our statisticians for their help with the statistical analysis performed in these experiments.
Glossary
Abbreviations
- BAL
broncho alveolar lavage
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- COPD
chronic obstructive pulmonary disease
- FEV100
forced expiratory volume in the first 100 milliseconds
- FITC
fluorescein isothiocyanate
- FVC
forced vital capacity
- GOLD
global initiative for chronic obstructive lung disease
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MUC5AC
muucin-5AC
- NaCMC
sodium carboxymethylcellulose
- NBF
neutral buffered formalin
- PBS
phosphate buffered saline
- PDE
phosphodiesterase
- PEG300
polyethylene glycol 300 <
- PRRs
Pathogen recognition receptors
- TLC
Total lung capacity
- TLR4
toll like receptor 4
- TS
tobacco smoke
- UEA-1
Ulex europaeus agglutinin-1
Conflict of interest
The authors have no conflict of interest.
References
- Aaron SD, Angel JB, Lunau M, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:349–355. doi: 10.1164/ajrccm.163.2.2003122. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]
- Alsaeedi A, Sin DD, McAlister FA. The effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review of randomized placebo-controlled trials. Am J Med. 2002;113:59–65. doi: 10.1016/s0002-9343(02)01143-9. [DOI] [PubMed] [Google Scholar]
- Baginski TK, Dabbagh K, Satjawatcharaphong C, Swinney DC. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am J Respir Cell Mol Biol. 2006;35:165–174. doi: 10.1165/rcmb.2005-0259OC. [DOI] [PubMed] [Google Scholar]
- Balbi B, Bason C, Balleari E, et al. Increased bronchoalveolar granulocytes and granulocyte/macrophage colony-stimulating factor during exacerbations of chronic bronchitis. Eur Respir J. 1997;10:846–850. [PubMed] [Google Scholar]
- Birrell MA, McCluskie K, Wong S, Donnelly LE, Barnes PJ, Belvisi MG. Resveratrol, an extract of red wine, inhibits lipopolysaccharide induced airway neutrophilia and inflammatory mediators through an NF-kappaB-independent mechanism. FASEB J. 2005;19:840–841. doi: 10.1096/fj.04-2691fje. [DOI] [PubMed] [Google Scholar]
- Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ. 2000;320:1297–1303. doi: 10.1136/bmj.320.7245.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley PM. COPD: what is the unmet need? Br J Pharmacol. 2008;155:487–493. doi: 10.1038/bjp.2008.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:154–161. doi: 10.1164/rccm.200610-1563OC. [DOI] [PubMed] [Google Scholar]
- Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthew JL, Barnes PJ. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:1635–1639. doi: 10.1164/ajrccm.160.5.9811058. 5 Pt 1. [DOI] [PubMed] [Google Scholar]
- Davies L, Angus RM, Calverley PM. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet. 1999;354:456–460. doi: 10.1016/s0140-6736(98)11326-0. [DOI] [PubMed] [Google Scholar]
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. doi: 10.1136/thorax.57.10.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost EM, Skwarski KM, Sauleda J, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60:293–300. doi: 10.1136/thx.2004.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, Yasuo M, Urushibata K, Hanaoka M, Koizumi T, Kubo K. Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur Respir J. 2005;25:640–646. doi: 10.1183/09031936.05.00047504. [DOI] [PubMed] [Google Scholar]
- Gaschler GJ, Skrtic M, Zavitz CC, et al. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med. 2009;179:666–675. doi: 10.1164/rccm.200808-1306OC. [DOI] [PubMed] [Google Scholar]
- Gompertz S, O'Brien C, Bayley DL, Hill SL, Stockley RA. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur Respir J. 2001;17:1112–1119. doi: 10.1183/09031936.01.99114901. [DOI] [PubMed] [Google Scholar]
- Grootendorst DC, Gauw SA, Verhoosel RM, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62:1081–1087. doi: 10.1136/thx.2006.075937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A, Kemp P, Giddings J, Sugar R. Development and validation of a lectin-based assay for the quantitation of rat respiratory mucin. Novartis Found Symp. 2002;248:94–105. [PubMed] [Google Scholar]
- Leclerc O, Lagente V, Planquois JM, et al. Involvement of MMP-12 and phosphodiesterase type 4 in cigarette smoke-induced inflammation in mice. Eur Respir J. 2006;27:1102–1109. doi: 10.1183/09031936.06.00076905. [DOI] [PubMed] [Google Scholar]
- Lee KM, Renne RA, Harbo SJ, Clark ML, Johnson RE, Gideon KM. 3-week inhalation exposure to cigarette smoke and/or lipopolysaccharide in AKR/J mice. Inhal Toxicol. 2007;19:23–35. doi: 10.1080/08958370600985784. [DOI] [PubMed] [Google Scholar]
- Meng QR, Gideon KM, Harbo SJ, et al. Gene expression profiling in lung tissues from mice exposed to cigarette smoke, lipopolysaccharide, or smoke plus lipopolysaccharide by inhalation. Inhal Toxicol. 2006;18:555–568. doi: 10.1080/08958370600686226. [DOI] [PubMed] [Google Scholar]
- Miravitlles M, Guerrero T, Mayordomo C, Sanchez-Agudo L, Nicolau F, Segu JL. Factors associated with increased risk of exacerbation and hospital admission in a cohort of ambulatory COPD patients: a multiple logistic regression analysis. The EOLO Study Group. Respiration. 2000;67:495–501. doi: 10.1159/000067462. [DOI] [PubMed] [Google Scholar]
- Miravitlles M, Calle M, varez-Gutierrez F, Gobartt E, Lopez F, Martin A. Exacerbations, hospital admissions and impaired health status in chronic obstructive pulmonary disease. Qual Life Res. 2006;15:471–480. doi: 10.1007/s11136-005-3215-y. [DOI] [PubMed] [Google Scholar]
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340:1941–1947. doi: 10.1056/NEJM199906243402502. [DOI] [PubMed] [Google Scholar]
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Lofdahl CG, Laitinen LA, et al. Long-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. European Respiratory Society Study on Chronic Obstructive Pulmonary Disease. N Engl J Med. 1999;340:1948–1953. doi: 10.1056/NEJM199906243402503. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Buist AS, Ma P, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: National Heart, Lung, and Blood Institute and World Health Organization Global Initiative for Chronic Obstructive Lung Disease (GOLD): executive summary. Respir Care. 2001;46:798–825. [PubMed] [Google Scholar]
- Rabe KF, Bateman ED, O'Donnell D, Witte S, Bredenbroker D, Bethke TD. Roflumilast – an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2005;366:563–571. doi: 10.1016/S0140-6736(05)67100-0. [DOI] [PubMed] [Google Scholar]
- Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:1418–1422. doi: 10.1164/ajrccm.157.5.9709032. 5 Pt 1. [DOI] [PubMed] [Google Scholar]
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608–1613. doi: 10.1164/ajrccm.161.5.9908022. [DOI] [PubMed] [Google Scholar]
- Sethi S, Murphy TF. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev. 2001;14:336–363. doi: 10.1128/CMR.14.2.336-363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SD, Ramsey SD, Lee TA. The economic burden of COPD. Chest. 2000;117(Suppl. 2):5S–9S. doi: 10.1378/chest.117.2_suppl.5s. [DOI] [PubMed] [Google Scholar]
- Tsoumakidou M, Tzanakis N, Chrysofakis G, Siafakas NM. Nitrosative stress, heme oxygenase-1 expression and airway inflammation during severe exacerbations of COPD. Chest. 2005;127:1911–1918. doi: 10.1378/chest.127.6.1911. [DOI] [PubMed] [Google Scholar]
- Underwood SL, Raeburn D, Lawrence C, Foster M, Webber S, Karlsson JA. RPR 106541, a novel, airways-selective glucocorticoid: effects against antigen-induced CD4+ T lymphocyte accumulation and cytokine gene expression in the Brown Norway rat lung. Br J Pharmacol. 1997;122:439–446. doi: 10.1038/sj.bjp.0701398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med. 1996;153:1530–1535. doi: 10.1164/ajrccm.153.5.8630597. [DOI] [PubMed] [Google Scholar]
- Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 1999;353:1819–1823. doi: 10.1016/s0140-6736(98)10019-3. [DOI] [PubMed] [Google Scholar]
- Wollin L, Bundschuh DS, Wohlsen A, Marx D, Beume R. Inhibition of airway hyperresponsiveness and pulmonary inflammation by roflumilast and other PDE4 inhibitors. Pulm Pharmacol Ther. 2006;19:343–352. doi: 10.1016/j.pupt.2005.09.002. [DOI] [PubMed] [Google Scholar]