

Abstract
BACKGROUND AND PURPOSE
Bladder cancer is a highly recurrent cancer after intravesical therapy, so new drugs are needed to treat this cancer. Hence, we investigated the anti-cancer activity of combretastatin A-4 (CA-4), an anti-tubulin agent, in human bladder cancer cells and in a murine orthotopic bladder tumour model.
EXPERIMENTAL APPROACH
Cytotoxicity of CA-4 was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, propidium iodide (PI) staining assay and clonogenic survival assay. In vivo microtubule assembly assay, cell cycle analyses, Western blot and cell migration assay were used to study the mechanism of CA-4. The effect of intravesical CA-4 therapy on the development of tumours was studied in the murine orthotopic bladder tumour model.
KEY RESULTS
CA-4 inhibited microtubule polymerization in vivo. Cytotoxic IC50 values of CA-4 in human bladder cancer cells were below 4 nM. Analyses of cell-cycle distribution showed CA-4 obviously induced G2-M phase arrest with sub-G1 formation. The analyses of apoptosis showed that CA-4 induced caspase-3 activation and decreased BubR1 and Bub3 in cancer cells. In addition to apoptosis, CA-4 was also found to induce the formation of multinucleated cells. CA-4 had a significantly reduced cell migration in vitro. Importantly, the in vivo study revealed that intravesical CA-4 therapy retarded the development of murine bladder tumours.
CONCLUSIONS AND IMPLICATIONS
These data demonstrate that CA-4 kills bladder cancer cells by inducing apoptosis and mitotic catastrophe. It inhibited cell migration in vitro and tumour growth in vivo. Hence, CA-4 intravesical therapy could provide another strategy for treating superficial bladder cancers.
Keywords: bladder cancer, combretastatin A-4, apoptosis, mitotic catastrophe, intravesical chemotherapy, murine orthotopic bladder tumour model
Introduction
Bladder cancer is reported to be the ninth most common type of cancer and the 13th most common cause of death by cancer worldwide (Parkin, 2008). In the United States, bladder cancer is also the third most common type of cancer in men and the 10th most common in women (Jemal et al., 2009). There are many risk factors involved in the formation of bladder tumours (Lower, 1982; Murta-Nascimento et al., 2007), including exposure to arylamines, smoking, low fluid intake, chronic urinary infection by Schistosomal haematobium, low N-acetyltransferase activity and the frequent use of hair dye. In Taiwan, urinary bladder cancer is the most common urological cancer today. As the population of smokers and users of hair dye is growing rapidly, the number of people suffering from bladder cancer is expected to rise year by year.
More than 90% of bladder cancers are transitional cell carcinoma (TCC). Approximately 80% of all TCC initially develop along the superficial papillary pathway; the other 20% that develop along the non-papillary pathway are at a high risk of progressing to muscle invasive disease with a substantial risk for the development of distant metastasis (Spiess and Czerniak, 2006). The superficial papillary TCC is usually managed with a transurethral resection, followed by intravesical administration of therapeutic agents, including bacille Calmette-Guérin (BCG), mitomycin-C, doxorubicin and so on (Barocas and Clark, 2008). Although intravesical BCG can prolong the length of progression-free survival after the cancers have been resected, long-term follow-ups have demonstrated that it seldom cures patients of bladder cancer (Gee et al., 2002). According to Dr Amling's report, half of superficial lesions recur after intravesical therapy, and as many as 10% to 30% progress to a higher grade and/or stage and form local invasive cancers (Amling, 2001). Therefore, there is a clear need for novel therapeutic agents for intravesical therapy that provide a better cure after transurethral resection.
Microtubules are hollow tubes consisting of α- and β-tubulin heterodimers, which play a critical role in a wide variety of eukaryotic cellular functions, including chromosome separation in mitosis, transportation of intracellular materials and the maintenance of the position of organelles (Mollinedo and Gajate, 2003). During mitosis, the microtubule is the critical molecule for the separation of chromosomes. The disruption of microtubule dynamics can induce cell cycle arrest at the M phase, formation of abnormal mitotic spindles and eventually trigger the apoptotic signal pathway (Wang et al., 1999). Based on this function, microtubule disruptors are usually quite lethal to cancer cells, which have a higher frequency of M entrance than normal cells. One microtubule disruptor, docetaxel, has been reported to be effective with no systemic toxicity observed in Phase I trial of superficial bladder cancer intravesical therapy (McKiernan et al., 2006). Docetaxel is a semi-synthetic analogue of paclitaxel and is a very expensive treatment for clinical breast cancer (Saloustros et al., 2008). Due to these reasons, other microtubule disruptors with lower costs have become attractive pharmacological candidates for bladder cancer intravesical therapy.
Combretastatin A-4 (CA-4), originally isolated from the South African Combretum caffrum tree, is an easily synthesized microtubule-destabilizing agent that inhibits microtubule assembly by binding with tubulin at the same site as that of colchicines (Pettit et al., 1989;1995a;). CA-4 induces microtubule disassembly resulting in a dramatic cytotoxic effect on various human cancer cells (Simoni et al., 2006). In vivo, CA-4 phosphate – the water soluble CA4 prodrug, that selectively targets endothelial cells, induces regression of nascent tumour neovessels, reduces tumour blood flow and causes central tumour necrosis – has already entered clinical trials (Tozer et al., 1999; Stevenson et al., 2003; Kanthou et al., 2004). These effects result from a disruption of the vascular endothelial cadherin/β-catenin/Akt signalling pathway, thereby leading to rapid vascular collapse and tumour necrosis (Vincent et al., 2005). In addition to targeting endothelial cells, CA-4 and CA-4 phosphate also induce mitotic catastrophe and cell death in human non-small lung cancer cells (Vitale et al., 2007), human gastric cancer cell lines (Lin et al., 2007) and human leukaemia (Petit et al., 2008). However, no study has yet focused on the effects of CA-4 on human bladder cancer cells.
In this study, we analysed the anti-cancer effect of CA-4 and studied its cellular mechanism in human bladder cancer cells. TSGH 8301 cells and BFTC 905 cells, two human bladder papillary transitional cell carcinomas isolated from two bladder cancer patients in Taiwan, were used for this study. The TSGH 8301 cell line was derived from a grade II/stage A bladder carcinoma from a 56-year-old Chinese man (Yeh et al., 1988), and the BFTC 905 cell line was from a grade II/stage C bladder carcinoma from a 51-year-old Chinese woman (Cheng et al., 1995). In addition, to evaluate the in vivo anti-tumour effects of intravesical CA-4, we set up a murine orthotopic bladder tumour model, a model most relevant to human bladder cancer (Black and Dinney, 2007).
Methods
Combretastatin A-4
The compound CA-4 (Figure 1A) was synthesized via the Wittig reaction method using triphenyl(3,4,5-trimethoxybenzyl)phosphonium and 4-methoxy-3-(tert-butyl-dimethylsiloxy)benzaldehyde to produce the protected cis CA-4, which was then desilylated with tetrabutylammonium fluoride to obtain the cis CA-4. The spectroscopic identification was analysed by NMR (Pettit et al., 1995b). According to HPLC analyses, the purity of CA-4 was higher than 98% (Figure 1A).
Figure 1.

Structure of CA-4 and its inhibitory effect on microtubule polymerization in vivo. (A) Chemical structure and HPLC analyses data of synthetic cis CA-4. (B and C) Inhibitory effect of CA-4 on microtubule polymerization in vivo. BFTC 905 (B) and TSGH 8301 (C) cells were treated with CA-4 or with the same volume of DMSO as control. After 6 h incubation, cells were lysed in lysis buffer. Cell lysates were centrifuged to separate polymerized microtubules from free tubulin dimers as described in Methods. β-Actin was used as a loading control. The columns represent the means from three independent experiments and presented as mean ± SD. *P < 0.05, a significant difference compared with the control cells. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide.
Reagents and antibodies
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Hoechst 33342, propidium iodide (PI), ribonuclease A (RNase A), Z-VAD-FMK, propylene glycol, crystal violet and anti-β-actin antibodies were purchased from Sigma (St. Louis, MO, USA). Anti-phospho-AKT (Thr308) was purchased from Santa Cruz (Santa Cruz, CA, USA). Anti-α-tubulin was purchased from Abcam (Cambridge, UK). Anti-MPM-2 was purchased from Upstate (Lake Placid, NY, USA). Anti-BubR1 and anti-Bub3 were purchased from BD Biosciences (San Jose, CA, USA). Anti-caspase-3 and anti-PARP were purchased from Cell Signaling Technology (Danvers, MA, USA). The millicell hanging cell culture inserts for the Transwell system were purchased from Millipore (Billerica, MA, USA). Peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, USA).
Cell culture
Human bladder papillary transitional cell carcinoma TSGH 8301 cells and BFTC 905 cells (Bioresource Collection and Research Center, Hsin Chu, Taiwan) were maintained in RPMI 1640 medium (Invitrogen Gibco, Carlsbad, CA, USA) supplied with 10% foetal bovine serum (FBS) (Biological Industries, M.P. Ashrat, Israel). Human uroepithelium cells SV-HUC-1 (Bioresource Collection and Research Center, Hsin Chu, Taiwan) were maintained in F-12 medium (Invitrogen Gibco, Carlsbad, CA, USA) supplied with 7% FBS. Mice bladder carcinoma MB49 cells were kindly provided by Dr Timonthy L. Ratliff (Purdue Cancer Center, West Lafayette, IN, USA). The MB49 cells were maintained in RPMI 1640 medium supplied with 10% FBS, 1% penicillin and 1% streptomycin. Cells were incubated in a CO2 incubator at 37°C, with 5% CO2 and 95% filtered air. For orthotopic bladder tumour implantation, MB49 cells were harvested by trypsinization and suspended in RPMI 1640 without FBS and antibiotics.
In vivo microtubule assembly assay
This method was performed as described by Kuo et al. (2004). Cells were initially seeded at 1 × 105 cells per well in 12-well dishes for 48 h and then treated with various concentrations of CA-4 or DMSO for 6 h. Cells then were washed with PBS three times before the addition of lysis buffer containing 20 mM Tris-HCl (pH 6.8), 1 mM MgCl2, 2 mM EGTA, 20 µg·mL−1 leupeptin, 0.5 mM PMSF, 1 mM vanadate, 2 µ g·mL−1 pepstatin A and 0.5% NP-40 for 15 min at room temperature. Pellets were collected after centrifugation at 13 200×g for 10 min at 4°C. The pellets were dissolved in SDS-PAGE sampling loading buffer and heated at 95°C for 10 min to dissolve the pellets. The resulting material was subjected to SDS-PAGE on 7.5% SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred to a PVDF membrane. Antibodies against α-tubulin were employed as the primary antibodies. Immunoblot analyses was carried out with secondary antibodies coupled to horseradish peroxidase. The enhanced chemiluminescence kit and VL Chemi-Smart 3000 were used for detection and quantification.
MTT assay
BFTC 905 and TSGH 8301 cells were initially seeded at 9 × 103 cells and SV-HUC-1 were seeded at 1.5 × 104 cells per well in 96-well dishes and then treated with various concentrations of CA-4 or DMSO for 48 h. After that, cell viability was determined by MTT assay, based on the conversion of the tetrazolium salt by mitochondrial dehydrogenase to a formazan product. After the cells had been incubated with MTT, the culture medium was discarded, and formazan products were dissolved in DMSO. Each well was measured by light absorbance at 490 nm.
Measurement of the cellular membrane integrity by PI staining assay
The cellular membrane integrity was detected by PI staining assay. Cells were cultured in 100-mm tissue-culture dishes for 24 h and then incubated with DMSO or CA-4 for the indicated time. After that, trypsinized cells were resuspended in PBS and stained for 10 min with 5 µg·mL−1 PI. Stained cells were excited by exposure to an argon laser at 488 nm, collection of fluorescence emission at 580 nm, and at least 10 000 cells counted with a Becton-Dickinson FACScan flow cytometer, using CellQuest software. Live cells deny the entrance of PI, indicating complete cellular membrane integrity; dead cells are stained by PI, indicating damage to the cellular membranes.
Clonogenic survival assay
The clonogenic survival assay is a well-established technique for determining cell proliferation capability (Kuo et al., 2006). To determine the long-term effects of transient drug exposure in cells, various concentrations of CA-4 were added to cultured cells for 1 h. After the cells had been rinsed with fresh medium, they were allowed to grow for 14 days to form colonies. After that, the cells were stained with crystal violet and photographed.
Cell cycle analyses
Both floating and adhesive cells were collected. Around 8 × 105 BFTC 905 cells, TSGH 8301 cells or 2 × 106 SV-HUC-1 cells were seeded in 100-mm dishes. After 24 h incubation for attachment, 10 nM CA-4 was added and cells were incubated for another 24 h and 48 h. After CA-4 treatment, cells were trypsinized, centrifuged and fixed with ice-cold 75% ethanol overnight at 4°C. After the ethanol had been removed, the cells were stained with a DNA staining solution (containing 1 mg·mL−1 PI and 10 mg·mL−1 RNase A dissolved in PBS) for 30 min at room temperature. The DNA content of the stained cells was measured using a FACScan flow cytometer. The cell doublets were removed by gating the left area of FL2-W/FL2-A plot for analyses. Cell cycle data from flow cytometry were analysed using ModFit LT™ software.
Measurement of multinucleated cells
Cells were initially seeded at 1 × 105 cells per well in 24-well dishes for 24 h and then treated with various concentrations of CA-4 for 24 h. At the end of the treatment, the cells were carefully washed with PBS. The nuclei were stained with 5 µg·mL−1 Hoechst 33342 for 30 min at 37°C. The morphology of multinuclei (two or more than two nuclei in one cell) was confirmed under a fluorescent microscope and compared with the observations obtained under the light field. The number of multinuclei cells was counted in randomly chosen microscope fields at 200× magnification. To calculate the percentage of multinucleated cells in one experiment at least 500 cells were examined.
In vitro cell migration assay
The 24-well plate Transwell system, with a polycarbonate filter membrane of 8-µm pore size, was used. Cells were seeded on the upper compartment of the Transwell chamber at a cell density of 2 × 105 in 200 µL RPMI-1640 medium for 24 h. The media in the upper chamber were replaced by serum-free RPMI-1640 media and various concentrations of CA-4 or DMSO, then the lower chamber was filled with 10% FBS-containing RPMI-1640 medium. After a 24 h incubation, the cells that remained on the upper surface of the filter membrane were removed, and the cells on the opposite surface of the filter membrane were stained with crystal violet for 30 s and photographed under microscopy at 100× magnification. The number of migrated cells was counted in five randomly chosen microscope fields.
Animals
Thirty female C57BL/6 mice aged five to 6 weeks were provided by the National Laboratory Animal Center (Taipei, Taiwan), and maintained at our animal care facility for 1 week prior to use. The mice were housed in polycarbonate cages (5 per cage), provided with food and water ad libitum and maintained on a 12–12 h light-dark cycle at 22 ± 2°C and 55 ± 20% relative humidity. The mice were divided into groups for two different treatments (vehicle only or CA-4 treatment). All experiments were approved by the Institutional Animal Care and Use Committee of National Chiayi University.
Murine orthotopic bladder tumour model and intravesical CA-4 therapy
The implantation of murine bladder cancer cells MB49 into C57BL/6 mice was carried out as reported previously (Chade et al., 2008) with slight modifications. Mice were anaesthetized by the i.p. administration of tiletamine/zolazepam solution at a dose of 30 mg/30 mg·kg−1 body weight. Subsequently, a 24-gauge Teflon intravenous catheter (BD) was inserted through the urethra into the bladder. To prepare the bladder for tumour implantation, a chemical lesion was induced in the bladder wall by intravesical instillation of 20 µL 0.1 M AgNO3. This produced an adequate and controlled diffuse cauterization of the bladder wall. After 10 s, the content was washed out by transurethral infusion of 1 mL PBS. Then, a suspension of 2 × 105 MB49 cells was instilled into the bladder. Mice were randomly assigned to two groups (15 mice per group), and three mice in each group were not implanted with a bladder tumour. Twenty-four hours after tumour implantation, intravesical vehicle or CA-4 therapy was initiated. One group was only treated with vehicle (23% DMSO and 30% propylene glycol in PBS) every other day, and the other group received 50 mg·kg−1 CA-4 every other day while the mice were anaesthetized. After the vehicle or CA-4 had been injected into the mice bladders, the external urethral orifice of each mouse was obstructed by attaching a small piece of 3M™ Micropore™ Surgical Tape for 1 h. After that, the tapes were removed, and the mice were returned to their original cages.
Statistic analyses
Numerical data are expressed as mean ± SD. Statistical evaluation was carried out by Student's t-test. In the animal study, differences among the data of vehicle and CA-4 treatment were analysed by one-way anova followed by Dunn's test.
Results
CA-4 inhibits microtubule polymerization in bladder cancer cells
Although CA-4 is a well-known microtubule polymerization blocker, we wanted to be certain whether or not CA-4 inhibited microtubule polymerization in human bladder cancer cells. To analyse the effect of CA-4 on microtubule assembly in vivo, we treated BFTC 905 and TSGH 8301 cells with various concentrations of CA-4 for 6 h. The results showed that microtubule polymerization was inhibited when the concentration of CA-4 reached 10 nM (Figure 1B and C). Because CA-4 is potentially converted to trans form by light, we also tested the effect of trans CA-4 on microtubule polymerization and cytotoxicity in TSGH 8301 cells. Trans CA-4 had no effect on microtubule polymerization (Figure S1) and cytotoxicity, even at 1000 nM. This suggests that only cis CA-4 inhibits intracellular microtubule polymerization and that trans CA-4 does not have this property. Therefore, we concluded that conversion of cis CA-4 to trans CA-4 by light will reduce its antitumour activity therefore it should be made sure that CA-4 is not exposed to light when it is being used as an anticancer drug in the clinic.
Cytotoxicity of CA-4 in two human bladder cancer cell lines and a normal uroepithelial cell line
The next experiment was conducted to evaluate the cytotoxic effect of CA-4 against two human bladder cancer cell lines and one SV40 virus-transformed uroepithelial cell line SV-HUC-1. From morphological observations, CA-4 was found to induce more than 80% of BFTC 905 and TSGH 8301 cells to detach from the dish, but this only occurred with about 50% of the SV-HUC-1 cells. Using the MTT assay, the results showed that CA-4 had a cytotoxic effect on human bladder cancer cells, BFTC 905 and TSGH 8301, with IC50 values between 2 nM and 4 nM (Figure 2A), and that the maximal inhibitory effect was about 80%. However, with the same treatment time, the maximal inhibitory effect of CA-4 on SV-HUC-1 was only 40% (Figure 2A). This suggests that urinary cancer cells are more sensitive to the effects of CA-4 than normal uroepithelium. To further identify whether CA-4-detached cells were alive or dead, the PI staining assay was applied. As shown in Figure 2B, the viability of the cells as assessed using the PI staining assay was higher than that determined with the MTT assay, indicating that some of the CA-4-detached BFTC 905 and TSGH 8301 cells were still alive. However, according to the results obtained with the PI staining assay, BFTC 905 and TSGH 8301 cells were more sensitive to the cytotoxic effects of CA-4 than the SV-HUC-1 cells (Figure 2B). In addition, the clonogenic survival assay was also applied to analyse the cytotoxic effect of transient CA-4 treatment in the two cancer cell lines. Even though CA-4 was only present in the medium transiently, at a high dose it inhibited the survival of the colonies of the BFTC 905 and TSGH 8301 cells (Figure 2C).
Figure 2.

(A) Cytotoxicity of CA-4 against two human bladder cancer cell lines and a normal human uroepithelium. BFTC 905 and TSGH 8301 cells were initially seeded at 9x103 cells per well and SV-HUC-1 was seeded at 1.5x104 cells per well in 96-well dishes and then treated with various concentrations of CA-4 or DMSO for 48 h. The cell viability was measured by MTT assay. Measurement was performed in quadruplicate and presented as mean ± SD. This experiment was repeated three times. (B) CA-4-induced cell death in two human bladder cancer cell lines and a normal human uroepithelium. BFTC 905 and TSGH 8301 cells were initially seeded at 8x105 cells and SV-HUC-1 was seeded at 2.8x106 cells in 100-mm dishes and then treated with various concentrations of CA-4 or DMSO for 24 h and 48 h. Then the cells were collected and stained with PI for flow cytometry analyses. The columns represent the means from three independent experiments and are presented as mean ± SD. (C) Effect of CA-4 on clonogenic survival assay. BFTC 905 and TSGH 8301 cells were initially seeded at 1x103 cells in 100-mm dishes and then treated with various concentrations of CA-4 or DMSO for 1 h. After being rinsed with fresh medium, cells were allowed to grow for 14 days to form colonies. The columns represent the means of triplicate experiment and are presented as mean ± SD. *P < 0.05, a significant difference compared with the control cells. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PI, propidium iodide.
Effect of CA-4 on the cell cycle
Next, we used a flow cytometer to examine the effect of CA-4 on cell cycle progression. As shown in Figure 3A, CA-4 had a dramatically effect on the cell cycle, inducing cell cycle arrest at the G2/M phase in both BFTC 905 and TSGH 8301 cells. However, we were unsure whether the arrest occurred specifically at the G2 or the M phase. Therefore, using MPM-2 antibodies, we further examined the status of phosphorylated polypeptides, which are found only in mitotic cells. After 24 h of CA-4 treatment, at concentrations ranging from 5 nM to 50 nM, a significant elevation of MPM-2 was detected (Figure 3B). The expression of MPM-2 confirmed the cells to be arrested in the M phase, not the G2 phase. In addition, CA-4 also induced sub-G1 formation by 7- to 25-fold in these two cancer cell lines (Figure 3A). The increase in CA-4-induced sub-G1 was higher in BFTC 905 cells than in TSGH 8301 cells. In normal human uroepithelium SV-HUC-1 cells, CA-4 also induced G2/M arrest (Figure 3C), but in these cells it only induced sub-G1 formation by two- to threefold. This provides further evidence that CA-4 has only a slight cytotoxic effect on SV-HUC-1 cells compared with TSGH 8301 and BFTC 905 cells.
Figure 3.


(A) Effect of CA-4 on cell cycle distribution in bladder cancer cells. BFTC 905 and TSGH 8301 cells were treated with 10 nM CA-4 or DMSO for 24 h or 48 h, then the cells were collected for flow cytometry cell cycle analyses. (B) The expression of MPM-2 after CA-4 treatment. BFTC 905 and TSGH 8301 cells were treated with various concentrations of CA-4 or DMSO for 24 h, then the cells were collected and lysed for Western blot. The columns represent the means from three independent experiments and are presented as mean ± SD. *P < 0.05, a significant difference compared with the control cells. (C) Effect of CA-4 on cell cycle distribution in SV-HUC-1 cells. Cells were treated with 10 nM CA-4 or DMSO for 24 h or 48 h, then the cells were collected for flow cytometry cell cycle analyses. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide.
CA-4 induces the biochemical features of apoptosis in bladder cancer cells
In Figure 3A, CA-4 is shown to induce different sub-G1 populations among the BFTC 905 and TSGH 8301 cells. As sub-G1 does not necessarily indicate apoptosis, as a further study, we tried to obtain other evidence of CA-4-induced apoptosis. Using the Western blotting technique, we found that CA-4 induced 66% of BFTC 905 cells, but only 35% of TSGH 8301 cells, to cleave and produce PARP and caspase-3 (Figure 4A, top). This indicates that CA-4 is more effective at inducing apoptosis in BFTC 905 cells than in TSGH 8301 cells. Therefore, we analysed an M phase check protein BubR1, which is required for M phase arrest and prevents apoptosis (Greene et al., 2008). BubR1 was highly expressed in TSGH 8301 cells, with only minor expression in BFTC 905 cells (Figure 4A, bottom). BubR1 began to be phosphorylated after 16 h of treatment with CA-4 in both cell lines, indicating that the same number of cells entered M phase at that time. BubR1 and Bub3 were dramatically reduced after 48 h of CA-4 treatment in BFTC 905 cells, but were only decreased by 10 to 20% in TSGH 8301 cells (Figure 4A, top). This suggests that under CA-4 treatment for 48 h, BFTC 905 cells could exit the M phase, but most of the TSGH 8301 cells stayed in the M phase. This might be why apoptotis was more easily identifiable in BFTC 905 cells than in TSGH 8301 cells. To understand the role of apoptosis in CA-4-treated cells, we further compared the effect of a pan-caspase inhibitor on CA-4-induced cell death in these two cell lines. Z-VAD-FMK 40µM reversed 61% and 45% of cell death in BFTC 905 and TSGH 8301 cells respectively (Figure 4B). Interestingly, Z-VAD-FMK did not completely inhibit cell death, but it did completely block CA-4-induced PARP and caspase-3 production in BFTC 905 cells (Figure 4C), indicating that there are other pathways leading to cell death in addition to CA-4-induced apoptosis.
Figure 4.
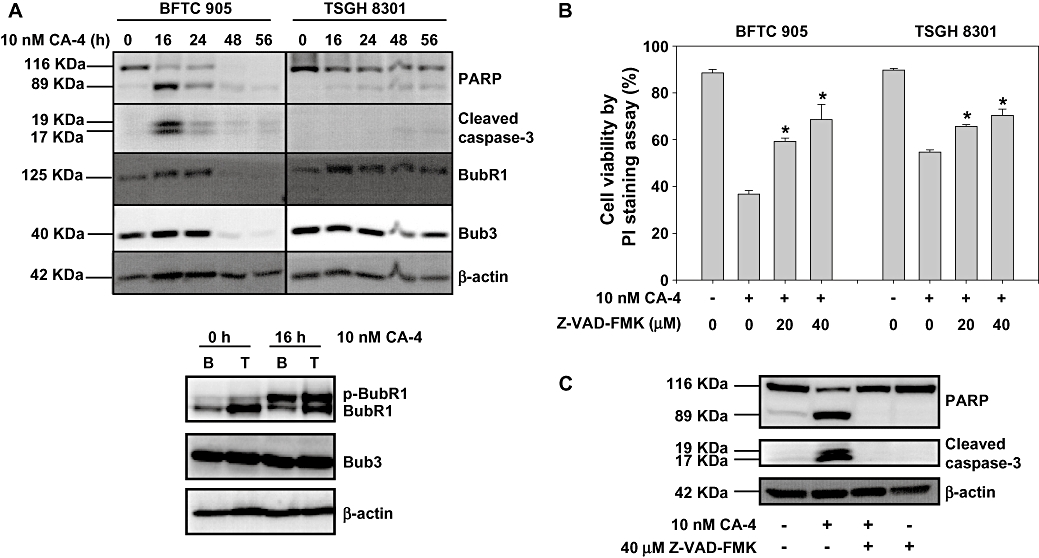
(A) Effect of CA-4 on the expression of apoptotic proteins and M phase checkpoint proteins. BFTC 905 (B) and TSGH 8301 (T) cells were treated with 10 nM CA-4 or DMSO for indicated times, then the cells were collected and lysed for Western blot. (B) Effect of Z-VAD-FMK on CA-4-induced cell death. BFTC 905 and TSGH 8301 cells were pretreated with or without different concentrations of Z-VAD-FMK for 1 h before being exposed to 10 nM CA-4 or DMSO for 24 h. The cell viability was measured by PI staining assay. The columns represent the means from three independent experiments and presented as mean ± SD. *P < 0.05, a significant difference compared with the CA-4-treated cells. (C) Effect of Z-VAD-FMK on CA-4-induced PARP and caspase 3 cleavage. BFTC 905 cells were pretreated with or without 40 µM Z-VAD-FMK for 1 h before being exposed to 10 nM CA-4 or DMSO for 24 h, then the cells were collected and lysed for Western blot. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide.
CA-4 induces the formation of multinucleated cells in bladder cancer cell lines
The formation of giant multinucleated cells is a phenotype of mitotic catastrophe induced by ionizing radiation or certain anticancer drugs (Castedo et al., 2004). We analysed whether CA-4 induced giant multinucleation in these two cell lines. As shown in Figure 5A, under the fluorescent field, the cells containing the enlarged multinucleated phenotype could be clearly observed after CA-4 treatment. The number of multinucleated cells was calculated by comparing those observed under the fluorescent field to those under the light field. CA-4 10 nM induced 30% and 18% of cells to become multinuclei in BFTC 905 and TSGH 8301 cells respectively (Figure 5B). In addition to the percentage of multinucleated cells of BFTC 905 being higher than that of TSGH 8301, the status of the multinucleated cells differed between the two cell lines. CA-4 induced more separated nuclei in BFTC 905 than in TSGH 8301, and more than two nuclei could be found only in BFTC 905 cells. This suggests that BFTC 905 cells could exit mitosis without cell division, but most of the TSGH8301 cells could not complete mitosis. Despite these differences, CA-4 indeed induced a mitotic catastrophe phenomenon in both bladder cancer cell lines.
Figure 5.


(A) CA-4 induced giant multinucleation in BFTC 905 and TSGH 8301 cells. Cells were treated with DMSO or 10 nM CA-4 for 24 h, and then stained with H33342 to label DNA. The light (left) and fluorescent (right) images of BFTC 905 and TSGH 8301 cells are presented. (B) Calculation of CA-4-induced multinucleation in BFTC 905 and TSGH 8301 cells. Representative light (left) and fluorescent (right) images of BFTC 905 and TSGH 8301 cells after 10 nM CA-4 treatment for 24 h. Arrows indicate binucleated or multinucleated cells. The scale bar 100 um indicates 100 micrometers. Lower panel shows tha number of multinucleated cells from three independent experiments. *P < 0.05, a significant difference between the control and the CA-4-treated cells. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide.
Effect of CA-4 on cell metastasis and AKT phosphorylation in bladder cancer cells
Figure 6A shows that a 24 h CA-4 treatment significantly decreased the migration of BFTC 905 and TSGH 8301 cells on the Transwell in a dose-response manner. AKT kinases have been implicated in various cellular responses, including tumour cell survival, proliferation, migration and invasiveness, and phosphorylated AKT is one of the most frequent molecules involved in the migration of tumour cells (Yoeli-Lerner and Toker, 2006). Hence, we investigated whether or not CA-4 inhibited the expression of p-AKT. In Figures 6B and 24 h CA-4 treatment decreased the expression of p-AKT in BFTC 905 cells, but not in TSGH 8301 cells. CA-4 reduced migration in both of these cancer cell lines, so its anti-metastasis effect was not only caused by inhibition of the expression of p-AKT.
Figure 6.


(A) Effect of CA-4 on cell migration. BFTC 905 or TSGH 8301 cells were seeded on the upper chamber of the transwell system for 24 h. The upper chamber media were replaced by serum-free media with various concentrations of CA-4 or DMSO, and the lower chamber was filled with 10% FBS media. After a 24 h incubation period, the cells remaining on the upper surface of the filter membrane were removed, and the cells on the opposite surface of the filter membrane were stained with crystal violet and photographed under microscopy. The histograms show the number of migrated cells. (B) Effect of CA-4 on AKT phosphorylation. Human bladder cancer cell lines were untreated or treated with various concentrations of CA-4 for 24 h. Cells were harvested and the cell extract was prepared and loaded on SDS-PAGE. β-Actin was used as a loading control. The means from three independent experiments are presented as mean ± SD in the histograms. *P < 0.05, a significant difference versus the control cells. CA-4, combretastatin A-4; DMSO, dimethylsulphoxide.
CA-4 retards tumour growth in the murine orthotopic bladder tumour model
To evaluate the inhibitory effect of CA-4 on bladder tumour progression in vivo, 30 mice with an orthotopic bladder tumour were used. Twenty-four mice were instilled with MB49 cells intravesically, and six mice were intravesically instilled with normal saline by a 24-gauge catheter. About 80% of the mice developed bladder tumours in this model (Table 1). Approximately 17% of the mice died due to penetration of the urethra caused by the intravesical technique; these mice all died the next day after the first intravesical application (the 2nd day). On the 13th day, the volume of the bladders was measured to examine the potential anti-tumour effect of intravesical therapy with CA-4. Figure 7B and C show that the mean bladder volume without tumour implantation was 10.6 ± 0.9 mm3. The bladder volume of the mice implanted with a tumour and intravesical vehicle was 96.2 ± 30.4 mm3. The mice implanted with tumour and intravesical CA-4 had bladder volumes of 33.6 ± 6.9 mm3 on the 13th day. The pathological section analyses showed that the transitional epithelium walls of the bladder in mice receiving CA-4 therapy without tumour implantation had no obvious lesions (Figure 7D-I). In the CA-4 treated, tumour-implanted mice, the tumour mass did not reach the muscle layer on the 13th day (Figure 7D-II), whereas in the group of mice implanted with a tumour and administered vehicle, the bladder carcinomas had spread out of the bladder connective tissue and muscle layer (Figure 7D-III), furthermore, the tumour thrombosis was also found in the mesenteric vessel (Figure 7D-IV). In addition to examining the histology of the bladder, the blood was also collected and analysed when the mice were killed (Table 2). The haematogram data showed that white blood cells and lymphocytes had decreased and platelets had increased after CA-4 treatment. No significant difference was found in the blood biochemical parameters of the blood from vehicle and CA-4 groups. Taken together, these results show that intravesical CA-4 did not damage normal urinary tissue, but it retarded the tumour growth significantly, and CA-4 might be absorbed from the bladder and affect the white blood cells and lymphocytes.
Table 1.
Orthotopic bladder tumour formation of mice with or without tumour implantation, vehicle or CA-4 treatment at termination of treatment
| Normal saline (n = 6) | MB49 (n = 24) | |||
|---|---|---|---|---|
| Installation treatment | Vehicle | CA-4 | Vehicle | CA-4 |
| No tumour | 3 | 2 | 0 | 1 |
| Tumour | 0 | 0 | 9 | 10 |
| No. of deaths | 0 | 1 | 3 | 1 |
CA-4, combretastatin A-4.
Figure 7.

Effect of CA-4 in a murine orthotopic bladder tumour model. (A) The time schedule for the murine orthotopic bladder tumour model. (B) The urinary system of mice with no tumour implantated and those treated with vehicle or CA-4 after tumour implantation. After the mice had been killed, the urinary system was isolated and photographed. Arrows indicate the bladders of mice with no tumour implanted (left), tumour-implanted mice treated with vehicle (middle) or treated with 50 mg·kg−1 CA-4 (right). ‘K’ indicates the kidney. (C) The bladder volume of a mouse with no tumour implanted and those with tumours treated with vehicle or CA-4. After the mice had been killed, the bladder volume was calculated by (length × width2)/2. *P < 0.05, a significant difference between the vehicle-treated and the CA-4-treated mice. (D) Bladder sections stained with haematoxylin and eosin (H&E) for histopathology. Magnification: 100x. (i) The bladder tissue of mice without a tumour after intravesical CA-4 therapy. ‘M’ stands for muscle layer. (ii) The bladder tissue of mice with a tumour and treated with 50 mg·kg−1 CA-4, the arrow indicates the edge of the tumour. (iii) The bladder tissue of mice with a tumour and treated with vehicle alone, the arrow indicates the outer edge of the bladder. (iv) The tumour in a vessel outside the bladder of the mice after tumour implantation and treatment with vehicle alone, the arrow indicates the tumour thrombosis. CA-4, combretastatin A-4.
Table 2.
Effect of vehicle or CA-4 on the haematograms and plasma biochemical parameters of mice at termination of treatment
| Vehicle | CA-4 | |
|---|---|---|
| Haematograms | ||
| WBC (×103·mm−3) | 8.17 ± 0.77 | 4.39 ± 0.48* |
| Ly (×103·mm−3) | 6.89 ± 0.65 | 3.49 ± 0.38* |
| RBC (×106·mm−3) | 7.47 ± 0.55 | 8.77 ± 0.43 |
| Hb (g·dL−1) | 8.84 ± 0.77 | 8.86 ± 0.54 |
| PLT (×103·mm−3) | 1093 ± 49.5 | 2861 ± 95.4* |
| Biochemical parameter | ||
| GOT (U·L−1) | 125.3 ± 23.1 | 130.3 ± 13.2 |
| GPT (U·L−1) | 24.2 ± 2.0 | 33.3 ± 1.4 |
| BUN (mg·dL−1) | 18.4 ± 2.1 | 19.0 ± 2.2 |
| Creatinine (mg·dL−1) | 0.5 ± 0 | 0.41 ± 0.02 |
| LDH (U·L−1) | 710 ± 134.9 | 494 ± 54.6 |
| CPK (U·L−1) | 420.5 ± 99.2 | 292.2 ± 53.1 |
Values are means ± SE.
P < 0.001, significant difference between vehicle and CA-4-treated mice.
WBC, white blood cell; Ly, lymphocyte; RBC, red blood cell; Hb, haemoglobin concentration; PLT, platelets; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; BUN, blood urea nitrogen; LDH, lactic dehydrogenase; CPK, creatine phosphokinase.
CA-4, combretastatin A-4.
Discussion and conclusions
Bladder cancer is the most common urological cancer in some countries, including Taiwan, where it is expected to rise year by year. Approximately 80% of all TCC initially develop as superficial papillary carcinoma, and it is usually managed with transurethral resection, followed by intravesical chemotherapy. Because the recurrent rate after intravesical chemotherapy is still high, we want to develop new agents to improve the intravesical chemotherapy. Here, we showed that CA-4 induces bladder cancer cell death through apoptosis and mitotic catastrophe, and inhibits cell migration in vitro. We also found that CA-4, when applied by the intravesical route, retards bladder tumour growth in vivo.
The molecular mechanisms by which CA-4 causes cell death are complex and are most likely mediated through caspase- and non-caspase-dependent pathways. Because CA-4 is a well-known microtubule-destabilizing agent that inhibits microtubule assembly (Dark et al., 1997; Kanthou et al., 2004), blocking microtubule dynamics will lead to apoptotic cell death (Mollinedo and Gajate, 2003). A caspase-independent mechanism by mitotic catastrophe has also been reported in the CA-4-induced death mechanism (Nabha et al., 2002). In the present study, we showed that CA-4 induces two different phenomena in BFTC 905 and TSGH 8301 cells. In the BFTC 905 cell line, CA-4 induced 66 to 90% of the cells to undergo PARP cleavage, caspase-3 activation and BubR1 degradation, but this only occurred in 10 to 35% of the TSGH 8301 cells. In spite of this difference, CA-4-induced cell death could be partially reversed by a pan-caspase inhibitor in both these cell lines, indicating that CA-4-induced cell death is partially mediated by the caspase pathway. CA-4 also induced the formation of giant multinucleated cells with more than two nuclei in BFTC 905 cells, but only slightly increased the size of cells to contain no more than two nuclei in the TSGH 8301 cell line. These multinucleated cells, and the abnormal separation of chromosomes, will lead to mitotic catastrophe and this effect may be used in anti-cancer therapy (de Bruin and Medema, 2008; Tao et al., 2009). Up to now, it has not been clear whether or not mitotic catastrophe activates non-caspase genome digestion, but non-caspase-induced genome digestion has been reported (Kim et al., 2008). From our data, non-caspase-induced cell death was shown to be an important mechanism in CA-4-induced cell death. The ability of CA-4 simultaneously to induce caspase- and non-caspase-dependent pathways has also been observed in leukaemic cells (Petit et al., 2008).
Combretastatin A-4 is highly potent at inducing cytotoxicity, and at inhibiting microtubule polymerization, and cell metastasis, because these effects are induced by concentrations lower than 100 nM. For both cytotoxicity and inhibition of microtubule polymerization, the maximum effect of CA-4 was obtained with 10 nM; this suggests that the cytotoxicity induced by CA-4 is predominantly mediated by blocking microtubule polymerization. Unlike cytotoxicity and inhibition of microtubule polymerization, the dose of CA-4 needed to inhibit cell metastasis did not reach a saturated effect at 10 nM, probably because there are many factors involved in cell metastasis and the mechanism of the anti-metastasis effect of CA-4 in these two cell lines has not been eluciadated. The potency of CA-4 for inhibiting microtubule polymerization and cell metastasis, in addition to its cytotoxicity after a 48 h treatment, as assessed by PI staining assay, were shown to be similar in the BFTC 905 and TSGH 8301 cells. This suggests that CA-4 has equal effects in these two bladder cancer cell lines at different stages of the tumour. Furthermore, a comparison of the bladder cancer cells and normal cells showed that CA-4 was less cytotoxic in SV-HUC-1 cells; because SV-HUC-1 is an SV-40 virus-transformed cell line, this immortalized cell line might provide unknown mechanisms for CA-4 resistance, but this requires further study. However, the pathology showed that intravesical CA-4 therapy in vivo also did not damage normal uroepithelium. This may be because CA-4 blocks microtubule dynamics that are more important for cancer cell division, which would make CA-4 more cytotoxic to cancer cells than normal cells.
Vincent et al. (2005) demonstrated that CA-4 inhibits endothelial cell migration and capillary tube formation through the disruption of the vascular endothelial-cadherin, β-catenin and AKT signalling pathway, thereby leading to rapid vascular collapse and tumour necrosis. A correlation between its anti-metastasis effect and p-AKT inhibition by CA-4 has also been reported in human gastric cancer cells (Lin et al., 2007). In this study, we found that CA-4 inhibited the migration of BFTC 905 and TSGH 8301 cells, but CA-4 was only found to inhibit p-AKT in BFTC 905 cells, not in TSGH 8301 cells. This implies that CA-4 inhibits the migration of bladder cancer cells in ways other than through a decrease in the expression of p-AKT. In fact, cell migration is regulated by various factors including the PI3K-AKT pathway (Yoeli-Lerner and Toker, 2006) and factors in epithelial-mesenchymal transitions (Thiery and Sleeman, 2006). In addition, CA-4 might also modulate cell migration by directly regulating the kinetics of tubulin, because there is strong interaction between the microtubules, intermediate filaments and actin filaments (Fuchs and Karakesisoglou, 2001), which are important for cell mobility. In vivo, there are more factors involved in cell metastasis, including matrix metalloproteinases (Roy et al., 2009) and angiogenesis (Folkman, 2002). CA-4 phosphate has been reported to inhibit the expression of hypoxia inducible factor 1α (Dachs et al., 2006), which might be involved in the anti-migration effects of CA-4 in vivo.
Combretastatin A-4 causes cell arrest at the M phase and MPM-2 expression in BFTC 905 and TSGH 8301 cells. Anti-MPM-2 was a monoclonal antibody originally raised against mitotic HeLa cells. Subsequently, it has been shown to specifically recognize a cell-cycle-regulated phosphoepitope present in mitotic and meiotic proteins from a wide variety of species (Davis et al., 1983). More than 50 different phosphorylated proteins are recognized by anti-MPM-2, including topoisomerase IIα, microtubule-associated proteins and protein kinase CK2 (Escargueil et al., 2000). MPM-2 epitope is also present in multiple kinases that might phosphorylate each other, indicating the complexity of the signalling pathways, which regulate mitotic entry (Renzi et al., 1997). CA-4-induced MPM-2 expression reached a plateau at 10 nM in both cells, suggesting this concentration is a saturated pharmacological dose for CA-4-induced M arrest in these two cells. In cell cycle analyses, the percentage of BFTC 905 cells in the M phase after a CA-4 48 h treatment (10.5%) was much lower than that after the CA-4 24 h treatment (64.5%). A similar result was found with both the TSGH 8301 and SV-HUC-1 cells. It might be due to the cell death from the M-arrested cells. We analysed the expression of MPM-2 in attached and floated SV-HUC-1 cells and this was found to be less after 48 h of CA-4 than after 24 h, especially in floated cells (Figure S2), which indicates that the floated cells were not at the M phase after CA-4 treatment for 48 h, but probably dead.
The major purpose of the murine orthotopic bladder tumour model is to analyse the in vivo effect of intravesical CA-4 therapy. In addition to inhibiting tumour growth in the bladder, CA-4 also reduced the numbers of white blood cells and lymphocytes, but increased the number of platelets, in murine blood. The dose used in this animal model might be high and the frequency of drug application might also be high therefore CA-4 might be absorbed from the bladder mucosa into the systemic blood. Myelosuppression was not found in a CA-4 phosphate single-dose Phase I study (Cooney et al., 2004), but neutropenia and thrombocytopenia occurred when CA-4 phosphate/carboplatin combination therapy was applied in advanced cancer patients (Bilenker et al., 2005). Therefore, CA-4 in the blood might also have affected the numbers of white blood cells and lymphocytes in our murine model. However, the increased platelet number might be a transient effect caused by a decrease in their rate of destruction by the spleen; this needs further study. In this animal model, the mice continued to die 2 weeks after cancer cell implantation without therapy therefore we chose to kill them after 13 days, and because the experimental period was only 13 days, the major aim of this murine orthotopic bladder tumour model is to analyse the tumour size and the haematological data, not distal metastasis. In fact, we also did not observe any enlarged lymph nodes in the abdomen or any other tumour in the liver, lung and abdominal cavity in these mice.
Combretastatin A-4 itself has not been used in clinical trials because of its poor water solubility, which makes it unsuitable for intravenous application. Therefore, a water-soluble form of CA-4 (CA-4 phosphate) has replaced CA-4 for clinical trials (Stevenson et al., 2003). According to the drug design study (Pettit et al., 1995b; Stevenson et al., 2003), CA-4 phosphate was rapidly converted to the active hydrophobic form of CA-4 by non-specific phosphatases in vivo. Intravesical therapy is a local administration method allowing direct contact of hydrophobic agents, and when the bladder tumour is at a superficial stage, the direct application of water-insoluble CA-4 might provide more effective anti-cancer effects than intravenously administered CA-4 phosphate.
In summary, our findings provide a mechanistic insight into the effects of CA-4 in two human bladder cancer cell lines, and a therapeutic evaluation of its intravesical application in vivo. Our data support the further evaluation of CA-4 as a potential agent for intravesical chemotherapy of superficial bladder cancer.
Acknowledgments
This work is supported by grants from the National Science Council NSC98-2320-B-415-002-MY3 of the Republic of China, and Chiayi Christian Hospital, Taiwan.
Glossary
Abbreviations
- BCG
bacille Calmette-Guérin
- CA-4
combretastatin A-4
- DMSO
dimethylsulphoxide
- DTT
DL-dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- FBS
foetal bovine serum
- IC50
50% inhibitory concentration
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NMR
nuclear magnetic resonance
- PBS
phosphate-buffered saline
- PI
propidium iodide
- RNase A
ribonuclease A
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- TCC
transitional cell carcinoma
Conflict of interest
The authors state no conflicts of interest.
Supporting information
Additional supporting information may be found in the online version of this article:
Figure S1 CA-4, but not trans CA-4, inhibits microtubule polymerization in TSGH 8301 cells. TSGH 8301 cells were treated with CA-4, trans CA-4 or DMSO for 6 h, then cell lysates were centrifuged to separate polymerized microtubules from free tubulin dimers as described in ‘Materials and Methods.’ The polymerized tubulins were analysed by Western blot.
Figure S2 MPM-2 expression after CA-4 treatment for 24 h and 48 h in SV-HUC-1 cells. SV-HUC-1 cells were treated with 10 nM CA-4 or DMSO for 24 h or 48 h, then the floated and attached cells were individually collected and lysed for Western blot. A: attached cells; F: floated cells.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Amling CL. Diagnosis and management of superficial bladder cancer. Curr Probl Cancer. 2001;25:219–278. doi: 10.1067/mcn.2001.117539. [DOI] [PubMed] [Google Scholar]
- Barocas DA, Clark PE. Bladder cancer. Curr Opin Oncol. 2008;20:307–314. doi: 10.1097/CCO.0b013e3282f8b03e. [DOI] [PubMed] [Google Scholar]
- Bilenker JH, Flaherty KT, Rosen M, Davis L, Gallagher M, Stevenson JP, et al. Phase I trial of combretastatin a-4 phosphate with carboplatin. Clin Cancer Res. 2005;11:1527–1533. doi: 10.1158/1078-0432.CCR-04-1434. [DOI] [PubMed] [Google Scholar]
- Black PC, Dinney CP. Bladder cancer angiogenesis and metastasis – translation from murine model to clinical trial. Cancer Metastasis Rev. 2007;26:623–634. doi: 10.1007/s10555-007-9084-9. [DOI] [PubMed] [Google Scholar]
- de Bruin EC, Medema JP. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat Rev. 2008;34:737–749. doi: 10.1016/j.ctrv.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Chade DC, Andrade PM, Borra RC, Leite KR, Andrade E, Villanova FE, et al. Histopathological characterization of a syngeneic orthotopic murine bladder cancer model. Int Braz J Urol. 2008;34:220–226. doi: 10.1590/s1677-55382008000200013. discussion 226–229. [DOI] [PubMed] [Google Scholar]
- Cheng YT, Li YL, Wu JD, Long SB, Tzai TS, Tzeng CC, et al. Overexpression of MDM-2 mRNA and mutation of the p53 tumor suppressor gene in bladder carcinoma cell lines. Mol Carcinog. 1995;13:173–181. doi: 10.1002/mc.2940130307. [DOI] [PubMed] [Google Scholar]
- Cooney MM, Radivoyevitch T, Dowlati A, Overmoyer B, Levitan N, Robertson K, et al. Cardiovascular safety profile of combretastatin a4 phosphate in a single-dose phase I study in patients with advanced cancer. Clin Cancer Res. 2004;10:96–100. doi: 10.1158/1078-0432.ccr-0364-3. [DOI] [PubMed] [Google Scholar]
- Dachs GU, Steele AJ, Coralli C, Kanthou C, Brooks AC, Gunningham SP, et al. Anti-vascular agent Combretastatin A-4-P modulates hypoxia inducible factor-1 and gene expression. BMC Cancer. 2006;6:280. doi: 10.1186/1471-2407-6-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997;57:1829–1834. [PubMed] [Google Scholar]
- Davis FM, Tsao TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci USA. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escargueil AE, Plisov SY, Filhol O, Cochet C, Larsen AK. Mitotic phosphorylation of DNA topoisomerase II alpha by protein kinase CK2 creates the MPM-2 phosphoepitope on Ser-1469. J Biol Chem. 2000;275:34710–34718. doi: 10.1074/jbc.M005179200. [DOI] [PubMed] [Google Scholar]
- Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Karakesisoglou I. Bridging cytoskeletal intersections. Genes Dev. 2001;15:1–14. doi: 10.1101/gad.861501. [DOI] [PubMed] [Google Scholar]
- Gee J, Sabichi AL, Grossman HB. Chemoprevention of superficial bladder cancer. Crit Rev Oncol Hematol. 2002;43:277–286. doi: 10.1016/s1040-8428(01)00190-1. [DOI] [PubMed] [Google Scholar]
- Greene LM, Campiani G, Lawler M, Williams DC, Zisterer DM. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzoxazepines. Mol Pharmacol. 2008;73:419–430. doi: 10.1124/mol.107.039024. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Kanthou C, Greco O, Stratford A, Cook I, Knight R, Benzakour O, et al. The tubulin-binding agent combretastatin A-4-phosphate arrests endothelial cells in mitosis and induces mitotic cell death. Am J Pathol. 2004;165:1401–1411. doi: 10.1016/S0002-9440(10)63398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Hsieh HP, Pan WY, Chen CP, Liou JP, Lee SJ, et al. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004;64:4621–4628. doi: 10.1158/0008-5472.CAN-03-3474. [DOI] [PubMed] [Google Scholar]
- Kuo PL, Hsu YL, Cho CY. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther. 2006;5:3209–3221. doi: 10.1158/1535-7163.MCT-06-0478. [DOI] [PubMed] [Google Scholar]
- Lin HL, Chiou SH, Wu CW, Lin WB, Chen LH, Yang YP, et al. Combretastatin A4-induced differential cytotoxicity and reduced metastatic ability by inhibition of AKT function in human gastric cancer cells. J Pharmacol Exp Ther. 2007;323:365–373. doi: 10.1124/jpet.107.124966. [DOI] [PubMed] [Google Scholar]
- Lower GM., Jr Concepts in causality: chemically induced human urinary bladder cancer. Cancer. 1982;49:1056–1066. doi: 10.1002/1097-0142(19820301)49:5<1056::aid-cncr2820490535>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- McKiernan JM, Masson P, Murphy AM, Goetzl M, Olsson CA, Petrylak DP, et al. Phase I trial of intravesical docetaxel in the management of superficial bladder cancer refractory to standard intravesical therapy. J Clin Oncol. 2006;24:3075–3080. doi: 10.1200/JCO.2005.03.1161. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- Murta-Nascimento C, Schmitz-Drager BJ, Zeegers MP, Steineck G, Kogevinas M, Real FX, et al. Epidemiology of urinary bladder cancer: from tumor development to patient's death. World J Urol. 2007;25:285–295. doi: 10.1007/s00345-007-0168-5. [DOI] [PubMed] [Google Scholar]
- Nabha SM, Mohammad RM, Dandashi MH, Coupaye-Gerard B, Aboukameel A, Pettit GR, et al. Combretastatin-A4 prodrug induces mitotic catastrophe in chronic lymphocytic leukemia cell line independent of caspase activation and poly(ADP-ribose) polymerase cleavage. Clin Cancer Res. 2002;8:2735–2741. [PubMed] [Google Scholar]
- Parkin DM. The global burden of urinary bladder cancer. Scand J Urol Nephrol. 2008;42(Suppl.):12–20. doi: 10.1080/03008880802285032. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Singh SB, Boyd MR, Hamel E, Pettit RK, Schmidt JM, et al. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1a) J Med Chem. 1995a;38:1666–1672. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Temple C, Jr, Narayanan VL, Varma R, Simpson MJ, Boyd MR, et al. Antineoplastic agents 322. synthesis of combretastatin A-4 prodrugs. Anticancer Drug Des. 1995b;10:299–309. [PubMed] [Google Scholar]
- Petit I, Karajannis MA, Vincent L, Young L, Butler J, Hooper AT, et al. The microtubule-targeting agent CA4P regresses leukemic xenografts by disrupting interaction with vascular cells and mitochondrial-dependent cell death. Blood. 2008;111:1951–1961. doi: 10.1182/blood-2007-05-089219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzi L, Gersch MS, Campbell MS, Wu L, Osmani SA, Gorbsky GJ. MPM-2 antibody-reactive phosphorylations can be created in detergent-extracted cells by kinetochore-bound and soluble kinases. J Cell Sci. 1997;110:2013–2025. doi: 10.1242/jcs.110.17.2013. Pt 17. [DOI] [PubMed] [Google Scholar]
- Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287–5297. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloustros E, Mavroudis D, Georgoulias V. Paclitaxel and docetaxel in the treatment of breast cancer. Expert Opin Pharmacother. 2008;9:2603–2616. doi: 10.1517/14656566.9.15.2603. [DOI] [PubMed] [Google Scholar]
- Simoni D, Romagnoli R, Baruchello R, Rondanin R, Rizzi M, Pavani MG, et al. Novel combretastatin analogues endowed with antitumor activity. J Med Chem. 2006;49:3143–3152. doi: 10.1021/jm0510732. [DOI] [PubMed] [Google Scholar]
- Spiess PE, Czerniak B. Dual-track pathway of bladder carcinogenesis: practical implications. Arch Pathol Lab Med. 2006;130:844–852. doi: 10.5858/2006-130-844-DPOBCP. [DOI] [PubMed] [Google Scholar]
- Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol. 2003;21:4428–4438. doi: 10.1200/JCO.2003.12.986. [DOI] [PubMed] [Google Scholar]
- Tao Y, Leteur C, Calderaro J, Girdler F, Zhang P, Frascogna V, et al. The aurora B kinase inhibitor AZD1152 sensitizes cancer cells to fractionated irradiation and induces mitotic catastrophe. Cell Cycle. 2009;8:3172–3181. doi: 10.4161/cc.8.19.9729. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, et al. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Res. 1999;59:1626–1634. [PubMed] [Google Scholar]
- Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, et al. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J Clin Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Antoccia A, Cenciarelli C, Crateri P, Meschini S, Arancia G, et al. Combretastatin CA-4 and combretastatin derivative induce mitotic catastrophe dependent on spindle checkpoint and caspase-3 activation in non-small cell lung cancer cells. Apoptosis. 2007;12:155–166. doi: 10.1007/s10495-006-0491-0. [DOI] [PubMed] [Google Scholar]
- Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol. 1999;44:355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- Yeh MY, Yu DS, Chen SC, Lin MS, Chang SY, Ma CP, et al. Establishment and characterization of a human urinary bladder carcinoma cell line (TSGH-8301) J Surg Oncol. 1988;37:177–184. doi: 10.1002/jso.2930370310. [DOI] [PubMed] [Google Scholar]
- Yoeli-Lerner M, Toker A. Akt/PKB signaling in cancer: a function in cell motility and invasion. Cell Cycle. 2006;5:603–605. doi: 10.4161/cc.5.6.2561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.