

Abstract
BACKGROUND AND PURPOSE
Gap junctions play important roles in the regulation of cell phenotype and in determining cell survival after various insults. Here, we investigated the role of gap junctions in aminoglycoside-induced injury to renal tubular cells.
EXPERIMENTAL APPROACH
Two tubular epithelial cell lines NRK-E52 and LLC-PK1 were compared for gap junction protein expression and function by immunofluorescent staining, Western blot and dye transfer assay. Cell viability after exposure to aminoglycosides was evaluated by WST assay. Gap junctions were modulated by transfection of the gap junction protein, connexin 43 (Cx43), use of Cx43 siRNA and gap junction inhibitors.
KEY RESULTS
NRK-E52 cells expressed abundant Cx43 and were functionally coupled by gap junctional intercellular communication (GJIC). Exposure of NRK-E52 cells to aminoglycosides, G418 and hygromycin, increased Cx43 phosphorylation and GJIC. The aminoglycosides also decreased cell viability that was prevented by gap junction inhibitors and Cx43 siRNA. LLC-PK1 cells were gap junction-deficient and resistant to aminoglycoside-induced cytotoxicity. Over-expression of a wild-type Cx43 converted LLC-PK1 cells to a drug-sensitive phenotype. The gap junction inhibitor α-glycyrrhetinic acid (α-GA) activated Akt in NRK-E52 cells. Inhibition of the Akt pathway enhanced cell toxicity to G418 and abolished the protective effects of α-GA. In addition, gentamycin-elicited cytotoxicity in NRK-E52 cells was also significantly attenuated by α-GA.
CONCLUSION AND IMPLICATIONS
Gap junctions contributed to the cytotoxic effects of aminoglycosides. Modulation of gap junctions could be a promising approach for prevention and treatment of aminoglycoside-induced renal tubular cell injury.
Keywords: gap junction, renal tubular epithelial cells, aminoglycoside, cytotoxicity, gap junctional intercellular communication, connexin 43, Akt, α-glycyrrhetinic acid
Introduction
The kidney is a major organ for filtration, secretion, re-absorption and ultimately excretion of drugs or drug metabolites. This makes it particularly vulnerable to several drugs. These drugs include aminoglycoside antibiotics (gentamycin), immunosuppressive agents (cyclosporin) and anti-tumour drugs (cis-diamminedichloroplatinum; cisplatin). Among them, the aminoglycosides are particularly well known for their cytotoxicity to renal tubular epithelial cells. Accumulation of aminoglycosides in renal tubular cells causes acute tubular cell necrosis, which accounts for significant morbidity after clinical use (Fillastre et al., 1983; Mingeot-Leclercq and Tulkens, 1999). These serious side effects markedly limit the use of the drugs in the clinic. It is therefore important to identify and characterize potential factors that control tubular cell susceptibility to aminoglycosides and to design novel preventive and therapeutic strategies.
Gap junctions are clusters of transmembrane channels that permit the direct intercellular exchange of ions, secondary messengers and small signalling molecules between cells. Intercellular communication via gap junction is thought to play an important role in the control of a variety of cellular functions, including cell growth, migration, differentiation and survival (Kumar and Gilula, 1996; Saez et al., 2003; Yao et al., 2007a; 2009;). Gap junctions are formed by a family of specialized proteins called connexins (Cx) and, so far, more than 20 different Cx proteins have been identified. Among them, Cx43 is predominantly expressed in a variety of cell types and has been extensively investigated. Recently, gap junctions have been shown to be critically involved in various pathological situations and these junctions can either attenuate or exacerbate the damage, depending on the properties and extent of injuries, as well as the nature of insults (Azzam et al., 2001; Huang et al., 2001; Krutovskikh et al., 2002; Nakase et al., 2003; 2004; Garcia-Dorado et al., 2004).
In vivo and in vitro studies have demonstrated the presence of gap junction proteins in a variety of renal cell types including proximal tubular epithelial cells (Hillis et al., 1997; Haefliger et al., 2001). However, information regarding gap junctions in the nephrotoxic effects of drugs is still lacking. Given that gap junctions are critically involved in the regulation of cell survival in various pathological situations, it was possible that gap junctions were involved in injury to renal tubular cells, induced by aminoglycosides. Here, we present evidence showing that gap junctions were up-regulated by aminoglycosides and contributed to the propagation of renal tubular cell injury. In addition, we identified the gap junction inhibitor α-glycyrrhetinic acid (α-GA) as a promising agent for the prevention and treatment of aminoglycoside-induced tubular cell injury.
Methods
Cell culture
Tubular proximal epithelial cell lines, NRK-E52 and LLC-PK1 (from the American Type Culture Collection, Rockville, MD, USA), were maintained in Dulbecco's modified Eagle's medium/F-12 (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 100 U mL−1 penicillin G, 100 µg mL−1 streptomycin, 0.25 µg mL−1 amphotericin B and 5% fetal bovine serum.
Measurement of gap junctional intercellular communication (GJIC)
GJIC was assessed by transfer of the membrane-impermeant fluorescent dye, Lucifer Yellow (LY), after single cell microinjection with an automated microinjection system (Zeiss Oberkochen, Jene, Germany) (Yao et al., 2000; 2002; 2005; 2006;).
Transfection of cells with wild-type Cx43
Wild-type Cx43-pEGFP1 (Cx43 labelled with enhanced green fluorescent protein) or control pEGFP-N1 vector were transfected into LLC-PK1 cells by using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instruction. For transient transfection experiment, cells were exposed to stimulants 24 h after transfection. To obtain LLC-PK1 cell clones stably expressing EGFP or WT-Cx43-EGFP, the selective medium containing 700 µg mL−1 G418 was added into the medium and renewed at 4 day interval. Clones with high levels of GFP or Cx43 were selected under the fluorescence microscope and used for this study.
Western blotting
Levels of Cx43 and Akt were evaluated by Western blot analysis as described before (Yao et al., 2000; 2005; 2006; 2007b;).
Immunocytochemistry
Immunocytochemical staining for Cx43 was performed by using an anti-Cx43 antibody as described previously (Yao et al., 2000; 2005; 2006; Yaoita et al., 2002).
Assessment of cell viability
NRK-E52 and LLC-PK1 cells in confluent culture were exposed to various stimuli for the indicated time intervals. Cell viability was evaluated by a WST-1 Assay Kit (MBL, Nagoya, Japan) following the instructions provided by the manufacturers.
Transient transfection of cells with siRNA
NRK cells were transiently transfected with siRNA specifically targeting Cx43 (Rn_Gja1-1 or 5_HP validated siRNA; Qiagen, Japan) or a negative control siRNA (AllStars Negative Control siRNA) at a final concentration of 20 nM using Hyperfect transfection reagent for 24 h. After that, cells were either left untreated or exposed to G418 or hygromycin for the indicated time. Cellular protein was extracted and subjected to Western blot analysis for Cx43. Cell viability was evaluated by WST assay.
Statistical analysis
WST assays were performed in quadruplicate, and data were expressed as means ± SE or SD. Statistical analysis was performed using one-way anova and the Dunnett test. Comparison of two populations was performed by Student's t-test. P < 0.05 was considered to be a statistically significant difference.
Materials
A wild-type Cx43-pEGFP expressing vector was a gift from Dr M. Oyamada (Department of Pathology and Cell Regulation, Kyoto Prefectural University of Medicine, Kyoto, Japan). This vector was constructed by ligation of the DraI fragment of rat Cx43 cDNA into the SmaI site of the pEGFP-N1 vector (Clontech, Palo Alto, CA, USA) (Oyamada et al., 2002). Anti-phospho-Akt and anti-Akt were purchased from Cell Signaling (Beverly, MA, USA). The selective Akt inhibitor (Akti-1/2) was obtained from Calbiochem (LaJolla, CA, USA). TITC-conjugated swine anti-rabbit immunoglobulin was obtained from DAKO (Glostoup, Denmark). All other reagents including anti-Cx43 and anti-β-actin antibodies were purchased from Sigma (St. Louis, MO, USA).
All nomenclature for the proteins and receptors used in the study follows Alexander et al. (2009).
Results
NRK-E52 and LLC-PK1 cells display different susceptibility to G418 and hygromycin
Gentamicin 418 (G418) and hygromycin are aminoglycoside antibiotics similar in structure and function to gentamycin. They are widely used for selection of eukaryotic cells that acquire corresponding resistance genes in transfection studies. During clone selection experiments, we accidentally noticed that two proximal tubular epithelial cell lines, NRK-E52 and LLC-PK1, displayed different susceptibility to these antibiotics. NRK-E52 cells were far more vulnerable than LLC-PK1 cells. As shown in Figure 1A and B, treatment of NRK cells with G418 or hygromycin for 48 h caused a concentration-dependent loss of cell viability, as evaluated by WST assay. However, they had little effects on LLC-PK1 cells.
Figure 1.
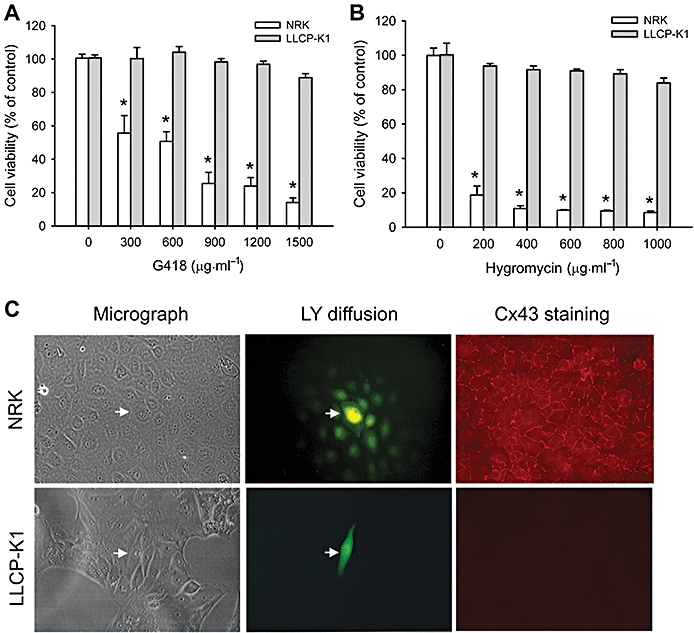
NRK-E52 and LLC-PK1 differ in response to aminoglycosides and in gap junction protein expression and function. (A and B) G418 and hygromycin induced cell injury in NRK-E52 and LLC-PK1 cells. NRK-E52 or LLC-PK1 cells in confluent cultures in 96-well plates were exposed to various concentrations of G418 or hygromycin for 48 h and cell viability was evaluated by WST assay. Data are expressed as percentage of living cells, compared with the untreated control. *P < 0.01 versus LLC-PK1 cells treated with respective G418. (C) Microscopic analysis of dye coupled cells and immunofluorescent staining of Cx43. Lucifer Yellow (LY) was pressure-injected into a single cell (arrow). Diffusion of LY into adjacent cells was recorded. Magnification, ×320. Expression of Cx43 was evaluated by staining the cells with an anti-Cx43 antibody. Magnification, ×400.
NRK-E52 and LLC-PK1 cells differ in gap junction expression and function
As gap junctions affect the response of cells to a range of stresses (Azzam et al., 2001; Huang et al., 2001; Krutovskikh et al., 2002; Nakase et al., 2003; 2004; Garcia-Dorado et al., 2004), we assessed the role of the gap junctions in the different cell responses to the aminoglycosides. To test this hypothesis, we first compared the expression and function of gap junctions in NRK-E52 and LLC-PK1 cells. Immunofluorescent staining revealed the presence of abundant gap junction protein Cx43 in NRK cells, which was localized at cell-to-cell contacts in a linear pattern. Moreover, the NRK-E52 cells had functional GJIC. Single cell injection of immunofluorescent dye, LY, led to diffusion of the dye from the injected cells to more than 10 surrounding cells (Figure 1C, upper panel). In contrast, LLC-PK1 cells had neither Cx43 nor GJIC (Figure 1C, lower panel). These results indicate that NRK-E52 and LLC-PK1 cells differed in the expression and function of gap junction proteins.
G418 promotes GJIC in NRK-E52 cells
We then examined whether aminoglycoside-induced NRK-E52 cell injury was preceded by altered gap junction expression and function. As shown in Figure 2A, incubation of NRK-E52 cells with G418 (100–500 µM) for 24 h caused morphological changes, manifested by the enlargement of lysosomes, a phenomenon that has been well described in gentamycin-injured tubular epithelial cells (Fillastre et al., 1983; Mingeot-Leclercq and Tulkens, 1999). This was associated with clearly altered Cx43 distribution and GJIC. In contrast to the punctate and linear distribution of Cx43 in control cells, Cx43 molecules in G418-treated cells were displayed at cell-to-cell contacts as aggregated particles (Figure 2B). Assay of LY dye transfer demonstrated that G418 treatment increased GJIC (Figure 2C and D). There were significantly more dye-coupled cells in G418-treated NRK cells than in control cultures. In addition, active dye transfer between morphologically normal and abnormal cells could be observed (Figure S1). Western blot analysis revealed that G418 promoted the conversion of Cx43 from a non-phosphorylated to a hyperphosphorylated form (Figure 2E). Similar findings were found in cells treated with hygromycin (Figure S2). These results indicate that aminoglycosides increased Cx43 phosphorylation and promoted GJIC in NRK-E52 cells.
Figure 2.
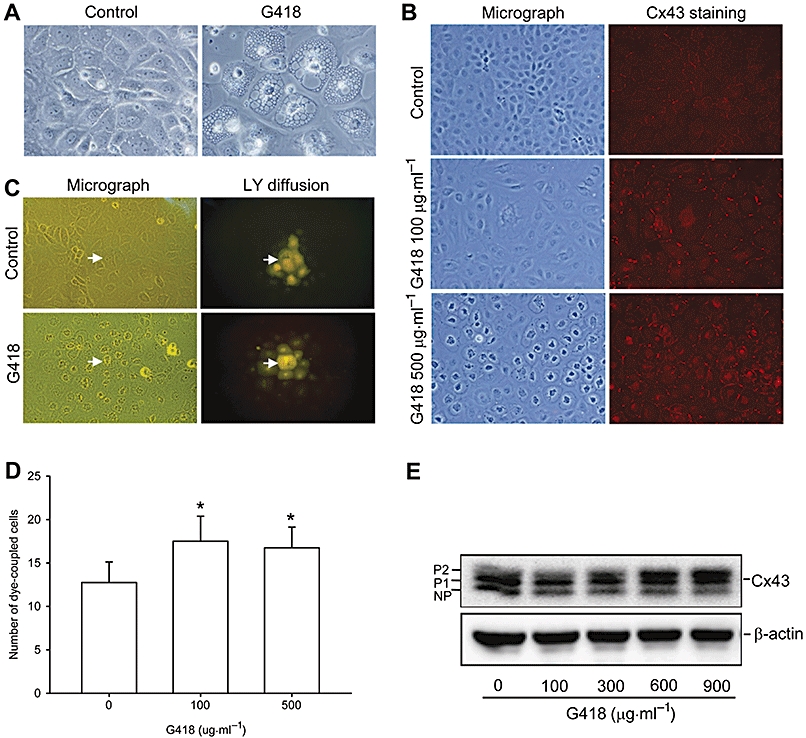
G418 elevates Cx43 expression and promotes GJIC in NRK-E52 cells. (A) Change of cell morphology in G418-treated NRK-E52 cells. NRK-E52 cells in confluent culture were either left untreated (control) or exposed to G418 (500 µg mL−1) for 24 h. Cell morphology was recorded by a CCD camera. Magnification, ×600. (B) Cx43 staining in G418-treated NRK-E52 cells. NRK-E52 cells in confluent culture were exposed to different concentrations of G418 for 24 h and subjected to immunofluorescent staining for Cx43. Magnification, ×400. (C) Functional GJIC in G418-treated cells. NRK-E52 cells in confluent culture were either left untreated (control) or exposed to G418 (500 µg mL−1) for 24 h. Lucifer Yellow (LY) was pressure-injected into a single cell. Diffusion of LY into adjacent cells was recorded by a CCD camera. Magnification, ×320. (D) Quantitative analysis of dye-coupled cells. The number of cells into which LY was transferred from the injected cells within 3 min is shown. Data are presented as the mean ± SD (n= 8), *P < 0.01 versus basal control. (E) Expression of Cx43 in G418-treated NRK-E52 cells. NRK-E52 cells were exposed to various concentrations of G418 for 24 h. Cellular proteins were extracted and subjected to Western blot analysis using an anti-Cx43 antibody. NP and P denote non-phosphorylated and phosphorylated Cx43, respectively. Equal loading of protein per lane was verified by reprobing the blots with an anti-β-actin antibody.
Gap junction inhibitors and Cx43 siRNA attenuate aminoglycoside-induced tubular cell injury in NRK-E52 cells
To examine whether the enhanced GJIC contributed to aminoglycoside-triggered cell injury, we measured cell viability in the presence or absence of gap junction inhibitors. First, we confirmed the effectiveness of the gap junction inhibitor α-GA in blocking GJIC. Exposure of NRK-E52 cells to α-GA (5 µM) for 30 min led to a rapid decrease of GJIC, as evaluated by the number of dye-coupled cells (P < 0.01; Figure 3A). Consistent with this finding, α-GA suppressed Cx43 protein levels, especially those of phosphorylated Cx43, as revealed by Western blot analysis (Figure 3B). Immunofluorescent staining demonstrated that the accumulation of Cx43 molecules at cell-to-cell contacts was greatly reduced in the presence of α-GA (Figure 3C).
Figure 3.
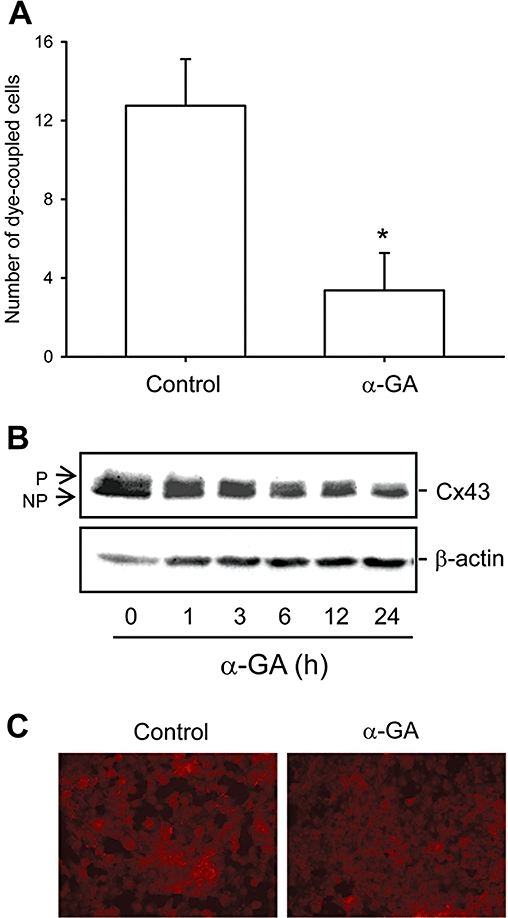
The gap junction inhibitor α-GA disrupts GJIC and decreases Cx43 protein levels. (A) Effect of α-GA on GJIC. NRK-E52 cells in confluent culture were either left untreated (control) or exposed to α-GA (5 µM) for 30 min. Lucifer Yellow (LY) was pressure-injected into a single cell. The number of cells into which LY was transferred from the injected cells within 3 min was counted. Data are presented as the mean ± SD (n= 8), *P < 0.01 versus basal control. (B) α-GA on Cx43 levels. NRK-E52 cells were treated with α-GA (5 µM) for the indicated times and cell protein was extracted and subjected to Western blot analysis by using an anti-Cx43 antibody. Equal loading of protein per lane was verified by reprobing the blot with an anti-β-actin antibody. (C) Immunofluorescent staining for Cx43 shows a decrease in NRK-E52 cells exposed to α-GA (5 µM) for 24 h. Magnification, ×200.
Using α-GA, we then evaluated the roles of gap junctions in aminoglycoside-induced cell injury. As shown in Figure 4A and B, α-GA attenuated the loss of cell viability induced by G418 in a concentration-dependent manner. This effect was mimicked by an active structural analogue of α-GA, carbenoxolone, that can disrupt GJIC, but not the inactive analogue, glycyrrhizic acid (GZA; Figure 4C) (Ozog et al., 2002). Furthermore, a similar effect was achieved by the structurally different gap junction inhibitors, heptanol and lindane (Figure 4D). Additionally, cytotoxicity induced by a clinically used aminoglycoside, gentamycin, in NRK-E52 cells was similarly prevented by α-GA and its active analogue carbenoxolone (Figure 4E).
Figure 4.
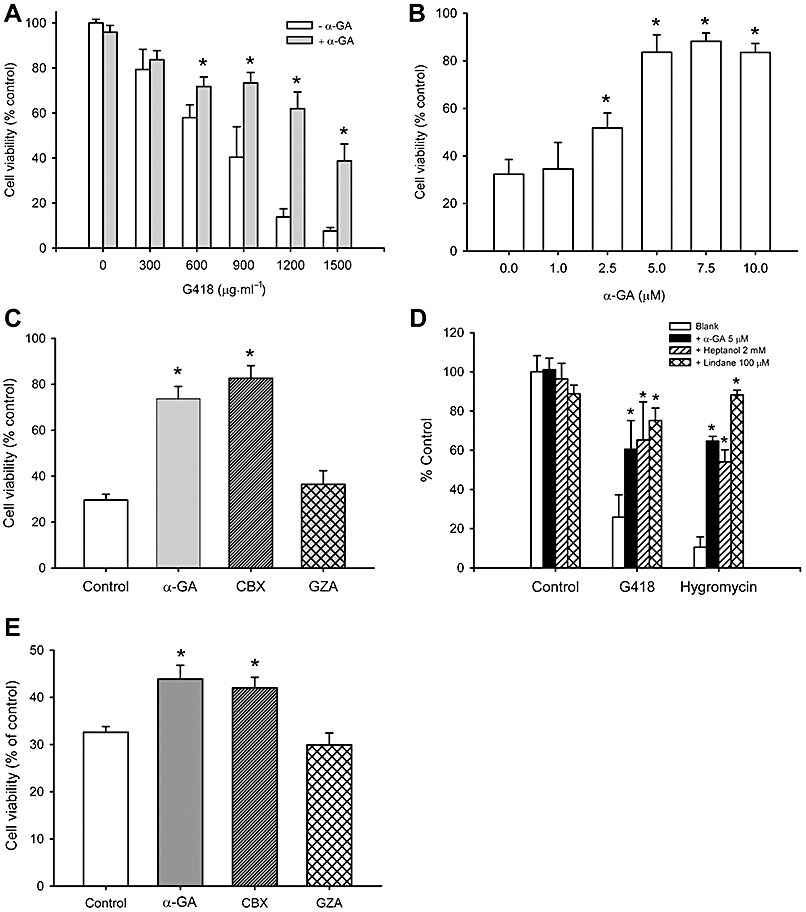
Gap junction inhibitors attenuate aminoglycosides-elicited cell injury in NRK-E52 cells. (A and B) Attenuation of drug-elicited cytotoxicity in NRK-E52 cells by α-GA. (A) NRK-E52 cells in confluent culture were exposed to various concentrations of G418 with or without α-GA (5 µM) for 48 h. (B) NRK-E52 cells were cultured with 1000 µg mL−1 G418 in the presence or absence of the indicated concentrations of α-GA for 48 h. (C) Effects of different analogues of α-GA on G418-elicited cytotoxicity. NRK-E52 cells were exposed to G418 (1000 µg mL−1) in the presence of 10 µM of α-GA, carbenoxolone (CBX), or glycyrrhizic acid (GZA) for 48 h. (D) Effect of different gap junction inhibitors on G418-elicited cytotoxicity. NRK-E52 cells were exposed to 1000 µg mL−1 G418 or 300 µg mL−1 hygromycin in the presence or absence of α-GA (5 µM), heptanol (2 mM) or lindane (100 µM) for 48 h. (E) Effects of different analogues of α-GA on gentamycin-elicited cytotoxicity. NRK-E52 cells were exposed to gentamycin (15 mg mL−1) in the presence of 10 µM of α-GA, CBX, or GZA for 48 h. Cell viability was evaluated by WST assay. Data are expressed as percentage of living cells, compared with the untreated control. *P < 0.01 versus untreated control.
The prevention by gap junction inhibitors of the cytotoxic effects of G-418 was also assessed by measurement of cell detachment or staining of cells with ethidium homodimer-2. These results are shown in Figures S3 and S4.
To exclude possible non-specific effects of gap junction inhibitors, we examined responses of NRK-E52 cells to G418 after down-regulation of Cx43 with siRNA. As shown in Figure 5A, Cx43 siRNA markedly suppressed Cx43 protein levels and significantly elevated cell resistance to hygromycin (Figure 5B) and G418 (Figure 5C). In contrast, control siRNA had no effects. These results indicate that gap junctions were critically involved in the cellular response to aminoglycosides.
Figure 5.
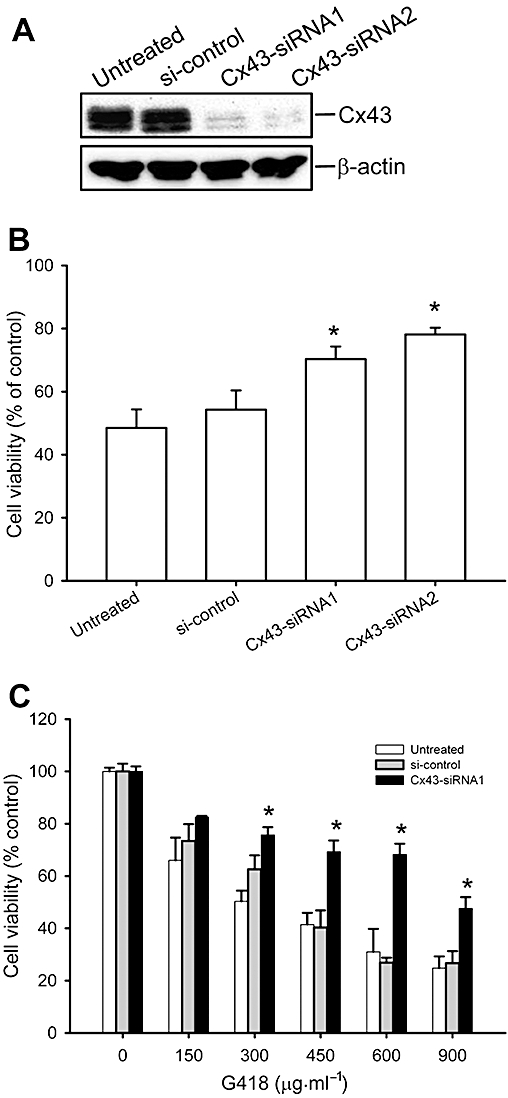
Cx43 siRNA attenuates aminoglycoside-elicited cell injury in NRK-E52 cells. (A) Down-regulation of Cx43 protein levels by Cx43 siRNA. NRK-E52 cells were transfected with Cx43-siRNA or control siRNA for 48 h. Cell proteins were extracted and subjected to Western blot analysis by using an anti-Cx43 antibody. Equal loading of protein per lane was verified by reprobing the blot with an anti-β-actin antibody. (B) Effect of Cx43 siRNA on hygromycin-induced cell injury. NRK-E52 cells were transfected with either Cx43 siRNA or control siRNA for 36 h and then exposed to hygromycin (150 µg mL−1) for an additional 48 h. (C) Effect of Cx43 siRNA on G418-induced cell injury. NRK-E52 cells were transfected with either Cx43 siRNA or control siRNA for 36 h and then exposed to the indicated concentrations of G418 for an additional 48 h. The cell viability was evaluated by WST assay. Data are expressed as percentage of living cells, compared with the untreated control. *P < 0.01 versus siRNA control.
Overexpression of Cx43 in LLC-PK1 cells sensitizes them to aminoglycoside-elicited cell injury
If gap junctions contribute significantly to drug-elicited tubular cell injury, one would expect that over-expression of Cx43 in the gap junction-deficient LLC-PK1 cells would sensitize them to the aminoglycosides. To test this possibility, LLC-PK1 cells were transfected with wild-type Cx43-EGFP or control pEGFP-N1 vector either transiently (Figure 6A) or permanently (Figure 6B). Because the vectors contain a neomycin phosphotransferase gene that makes the transfectants resistant to G418, we only examined the cytotoxic effect of hygromycin in these experiments. As shown in Figure 6, LLC-PK1 cells positively transfected with wild-type Cx43-EGFP showed plaque-like or linear localization of the fusion protein on the plasma membrane between adjacent cells (micrographs). These cells exhibited a much higher sensitivity to hygromycin than control cells transfected with a control plasmid (pEGFP-N1).
Figure 6.
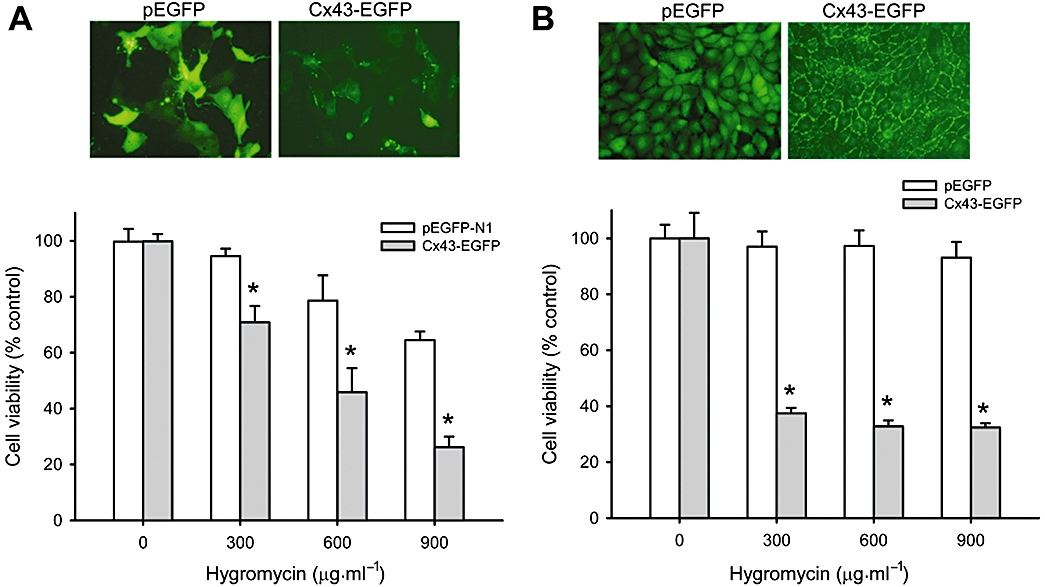
Over-expression of wild-type Cx43 in LLC-PK1 renders them vulnerable to hygromycin. (A) Transient transfection of LLC-PK1 cells with wild-type Cx43 on hygromycin-elicited cytotoxicity. LLC-PK1 cells were transiently transfected with a vector encoding GFP protein (pEGFP) or Cx43 (Cx43-EGFP) and cell response to G418 was evaluated by exposing the cells to different concentrations of hygromycin for 48 h. The expression of GFP and Cx43 is shown in the micrographs above the bar graphs. (B) Permanent transfection of LLC-PK1 cells with wild-type Cx43 on hygromycin-elicited cytotoxicity. LLC-PK1 cells were transfected with pEGFP or Cx43-EGFP and clones expressing a high level of GFP and Cx43 were selected, and analyzed for the cell response to hygromycin. The expression of GFP and Cx43 is shown in the micrographs above the bar graphs. Note the linear distribution of Cx43-GFP at the regions of cell-to-cell contacts. Magnification, ×400. *P < 0.01 versus respective control.
Akt activation contributes to the cytoprotective effects of the gap junction inhibitor α-GA
Because Akt is one of the well-known signal molecules involved in drug and apoptosis resistance (Krystal et al., 2002; Vivanco and Sawyers, 2002; Chung et al., 2006), we tested the contribution of Akt activation to the protective effects of the gap junction inhibitors. As shown in Figure 7A, exposure of NRK-E52 cells to α-GA resulted in a time-dependent activation of Akt. This action of α-GA was independent of G418, and could be mimicked by the active structural analogue carbenoxolone, but not the inactive analogue, GZA (Figure 7B).
Figure 7.
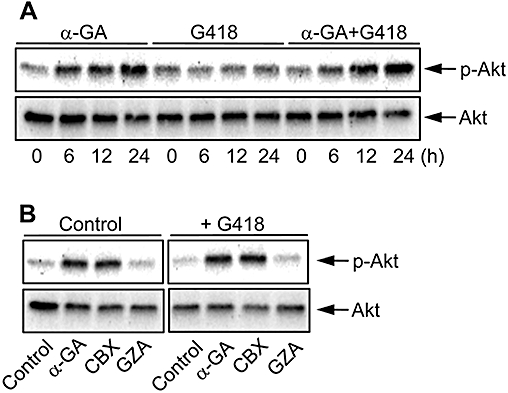
The α-GA activates Akt in NRK-E52 cells. (A) Activation of Akt by α-GA. NRK-E52 cells in confluent culture were exposed to α-GA (10 µM) in the presence or absence of G418 (500 µg mL−1) for the indicated time intervals. Cell proteins were extracted and subjected to Western blot analysis using an anti-phospho-Ser473-Akt-specific antibody (upper panel) or anti-Akt antibody (lower panel). (B) Effects of different analogues of α-GA on Akt activation. NRK-E52 cells were exposed to G418 (500 µg mL−1) in the presence of 10 µM of α-GA, carbenoxolone (CBX) or glycyrrhizic acid (GZA) for 24 h.
To evaluate the role of Akt activation in the cytoprotective effects of the gap junction inhibitors, we next examined cell responses to G418 after blocking the phosphatidyl 3-kinase (PI3K)/Akt pathway. As shown in Figure 8A and B, inhibition of PI3K with LY294002 or wortmanin, or inhibition of Akt with the selective Akt inhibitor, Akti-1/2, clearly sensitized cells to the cytotoxic effects of G418. These results indicate that the PI3K/Akt pathway functioned as a survival mechanism for NRK-E52 cells against G418-elicited cell injury.
Figure 8.
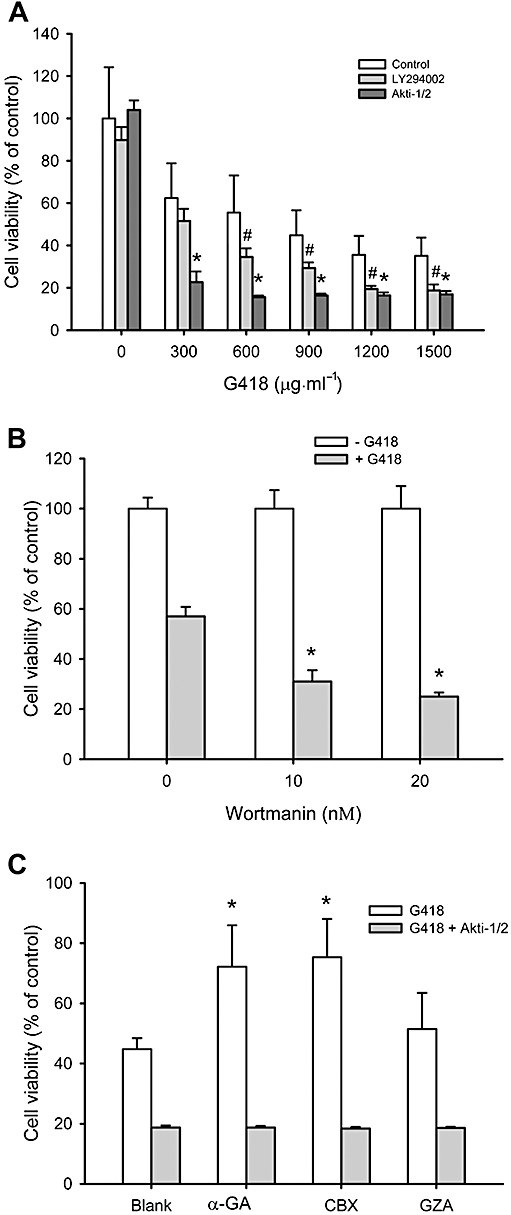
The PI3K/Akt pathway protects NRK-E52 cells from G418-elicited cell injury. (A and B) Effect of PI3K/Akt inhibition on G418-elicited cell injury. (A) NRK-E52 cells in confluent culture were exposed to the indicated concentrations of G418 in the presence or absence of LY294002 (50 µM) or Akti-1/2 (10 µM) for 48 h. (B) NRK-E52 cells were treated with 500 µg mL−1 G418 in the presence of the indicated concentrations of wortmannin for 48 h. The cell viability was evaluated by MTT assay. Data are expressed as percentage of living cells, compared with the control. # and *P < 0.05 versus cells treated with G418 alone. (C) Influence of Akt inhibition on the protective effect of α-GA and carbenoxolone (CBX). NRK-E52 cells were exposed to G418 (1000 µg mL−1) in the presence of α-GA, CBX or glycyrrhizic acid (GZA) (all 10 µM) with or without Akti-1/2 (10 µM) for 48 h. Cell viability was evaluated by WST assay. Data are expressed as percentage of living cells, compared with the untreated control. *P < 0.01 versus untreated control.
To show a direct involvement of Akt in the protective effects of gap junction inhibitors, we examined cell viability in the presence of the Akt inhibitor. As shown in Figure 8C, inhibition of Akt with Akti-1/2 completely abolished the cell protective actions of α-GA and carbenoxolone on G418-elicited cytotoxicity in NRK-E52 cells.
Discussion and conclusions
In this study, we demonstrated that gap junctions were one of the major determinants governing the susceptibility of renal tubular cell to aminoglycosides. This conclusion was supported by several observations. First, gap junction-rich NRK-E52 cells were found to be much more sensitive to aminoglycosides G418 and hygromycin than gap junction-deficient LLC-PK1 cells. Second, treatment of NRK-E52 cells with G418 or hygromycin increased Cx43 phosphorylation and promoted GJIC. Third, down-regulation of Cx43 or disruption of GJIC in NRK-E52 cells with gap junction inhibitors or Cx43 siRNA significantly attenuated aminoglycoside-initiated cell injury. Lastly, over-expression of a wild-type Cx43 in gap junction-deficient LLC-PK1 cells converted them to an aminoglycoside-sensitive phenotype.
The precise mechanisms by which gap junctions regulated cell susceptibility to aminoglycosides are presently unclear. In tumour therapy, gap junction-mediated bystander cell killing have been extensively documented. Gap junctions can transfer the toxic metabolites of ganciclovir from HSV-TK-expressing cells to the surrounding cells that do not express it (Dilber et al., 1997; Marconi et al., 2000). Gap junctions also transmit damage signals from irradiated to non-irradiated cells (Azzam et al., 2001). In addition, apoptotic and inflammatory signals can also pass through gap junctions (Krutovskikh et al., 2002; Krysko et al., 2004; Parthasarathi et al., 2006). In vivo studies have shown that gap junctions were critically involved in the propagation of ischaemic cell injury (Garcia-Dorado et al., 2004). Similarly, gap junction-mediated amplification of renal tubular cell in this study could also be ascribed to the intercellular spread of ‘toxic signals’. In support of this speculation, G418-initiated cell death was preceded by elevated GJIC. In addition, direct communication between morphologically normal and abnormal cells was observed. Furthermore, the gap junction inhibitor heptanol that disrupts intercellular communication without affecting Cx43 (Takens-Kwak et al., 1992; Cronier et al., 2003) also attenuated cell injury. All these pieces of evidence point to an involvement of the communication-dependent mechanism in the propagation of the renal cell injury. The nature of the signal molecules transmitted by the gap junctions remains to be characterized. The possible candidates may include superoxide and calcium ions. These molecules have been described to be able to pass through gap junctions to propagate toxic responses in several different pathological situations (Lin et al., 1998; Azzam et al., 2001; Krutovskikh et al., 2002). Moreover, production of superoxide and induction of Ca2+ release from endoplasmic reticulum by aminoglycosides have been previously reported (Jin et al., 2004; Sha and Schacht, 1999).
Gap junctions also have communication-independent actions (Zhang et al., 2003). For example, transfection of glioblastoma tumours with a wild-type Cx43 increased cell susceptibility to chemotherapeutic drugs, presumably through the communication-independent down-regulation of anti-apoptotic Bcl2 levels (Huang et al., 2001). However, the participation of this pathway in the current investigation was less likely. Our preliminary studies demonstrated that transfection of LLC-PK1 cells with a communication-free, mutant, Cx43 did not alter the cell response to hygromycin (Figure S5). In addition, the gap junction inhibitor heptanol that disrupts GJIC through decreasing membrane fluidity, without affecting Cx43 (Takens-Kwak et al., 1992; Cronier et al., 2003), also prevented G418-elicited cell death.
The effect of Cx43 on the increased cell susceptibility to aminoglycoside could also result from an increase in drug uptake and retention, and/or decreased elimination of the drugs. Indeed, the gap junction protein Cx32 enhanced vinblastine-elicited cytotoxic effects in renal carcinoma cells through suppression of a multidrug resistant gene-1 product, P-glycoprotein (an energy-dependent transport pump capable of decreasing the intracellular drug concentrations) (Sato et al., 2007). The participation of similar mechanisms in the current study is likely and more detailed studies of this aspect will be needed in the future.
In the present investigation, we found that the gap junction inhibitor α-GA could significantly prevent the cytotoxic effects of aminoglycosides on NRK-E52 cells. This effect of α-GA must be due to its action on gap junctions, because a similar protection was also provided by Cx43 siRNA and by other gap junction inhibitors. In addition, as found in type II alveolar cells (Guo et al., 1999), α-GA disrupted GJIC and decreased Cx43 levels, especially the phosphorylated form of Cx43 in NRK cells. Interestingly, α-GA also activated the Akt pathway, a major survival mechanism that is closely associated with drug and apoptosis resistance in various cell types (Vivanco and Sawyers, 2002; Chung et al., 2006; Kuwana et al., 2008; Meier et al., 2008). An earlier report indicated that the inhibition of Akt sensitized tumour cells to traditional chemotherapeutic agents (Krystal et al., 2002) and we found that the inhibition of Akt exacerbated G418-elicited cell injury and abolished the protective effect of α-GA. At present, the mechanisms responsible for Akt activation and relationship between Akt activation and gap junction disruption by α-GA are still unclear. Considering the important roles of Akt activation in cell survival, activation of Akt could be a novel mechanism contributing to the cytoprotective effects of α-GA.
Another novel finding in this study is that the aminoglycosides increased GJIC and increased Cx43 phosphorylation in NRK-E52 cells. The mechanisms involved are still unclear. One possibility is that aminoglycosides regulates Cx43 via interfering with Cx43 degradation. Aminoglycosides have been reported to induce enlargement of lysosomes and the suppression of several lysosomal enzymes (Fillastre et al., 1983; Mingeot-Leclercq and Tulkens, 1999). As the lysosome is one of the major organelles involved in Cx43 degradation (Qin et al., 2003), inhibition of this organelle could lead to Cx43 accumulation and hence, increased GJIC (Guan and Ruch, 1996).
It is worth noting that the aminoglycosides induced phosphorylation of Cx43, a molecular event that has been shown to be closely related to the functional status of gap junctions (Laird, 2005; Lau, 2005; Solan and Lampe, 2009). Usually, increased Cx43 phosphorylation is associated with a decrease of GJIC (Laird, 2005; Lau, 2005; Pahujaa et al., 2007). However, there are a growing number of publications with different results (Imanaga et al., 2004; Laird, 2005; Lampe et al., 2006; Procida et al., 2009; Shintani-Ishida et al., 2009). For example, phosphorylation of Cx43 by protein kinase A was associated with an elevated GJIC (Imanaga et al., 2004). At present, the reasons for this divergence in results are still unclear. One explanation is that Cx43 molecule has multiple tyrosine and serine sites and phosphorylating different sites could have different effects on GJIC. Thus, phosphorylation of Cx43 at serine 368 by PKC decreased GJIC (Lampe et al., 2000), whereas phosphorylation of Cx43 at serine residues 306, 325, 328 and/or 330 of Cx43 increased GJIC (Lampe et al., 2006; Procida et al., 2009). Therefore, in this study, it is likely that an elevated Cx43 phosphorylation could lead to an enhanced GJIC. At present, the signalling mechanisms involved in Cx43 phosphorylation and the functional role of phosphorylated Cx43 in aminoglycoside-elicited cell injury are still unclear. Further studies are needed to clarify these points.
Our findings may have important clinical implications. Renal tubular epithelial cells express several gap junction proteins including Cx43 (Hillis et al., 1997; Haefliger et al., 2001). However, the pathological roles of these proteins remain obscure. Using a cell culture model, we have characterized gap junctions as a hitherto unrecognized factor controlling tubular cell responses to aminoglycosides. Regulation of gap junctions could be an important mechanism contributing to the cytotoxic effects of aminoglycoside. In addition, we identified α-GA as a promising agent for prevention and treatment of the drug-initiated renal tubular cell injuries. The α-GA could not only restrict the propagation of ‘toxic signal’ through disruption of gap junctions, but also enhance cell resistance to the toxic agents through activation of the cell survival Akt signalling pathway. It remains to be determined whether our in vitro findings could be confirmed in vivo. This will be the focus of our future investigation. Additionally, our findings may explain why cells of different types vary in their responses to G418 and hygromycin during clone selection. It appeared that the regulatory effects of gap junction on cell sensitivity to drugs were not cell-type specific. The similar protective effects of α-GA on G418-elicited cell damage could be observed in mesangial cells (data not shown). Furthermore, a close correlation between Cx43 levels and cell susceptibility to G418 was established by using fetal fibroblasts derived from Cx43 wild-type (Cx43+/+), heterozygous (Cx43±) and knockout (Cx43−/−) littermates (Figure S6).
In conclusion, we have demonstrated that gap junctions control the susceptibility of renal tubular cells to aminoglycosides. Regulation of gap junctions could be a novel mechanism implicated in the cytotoxic effects of some drugs. Targeting gap junctions could be a novel and promising strategy against drug-elicited cell injury.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (17659255 and 20590953 to J. Y. and B21390314 to H. M.) and grants from Takeda Science Foundation, Japan-China Medical Association as well as Strategic Project grant from University of Yamanashi. We thank Dr M. Oyamada for the generous gift of Cx43-pEGFP expressing vector.
Glossary
Abbreviations
- α-GA
α-glycyrrhetinic acid
- Cx
connexin
- G418
gentamicin 418
- GJIC
gap junctional intercellular communication
- GZA
glycyrrhizic acid
- LY
Lucifer Yellow
- PKA
protein kinase A
Statement of conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Dye transfer between morphologically normal and abnormal NRK-E52 cells. NRK-E52 cells were exposed to 500 μg·mL−1 G418 for 24 h. A scrape line on the monolayer was made with a surgical blade. Lucifer Yellow (LY) was added and allowed for diffusion for 3 min. After washing out background fluorescence, the cells were fixed and photographed with a digital camera attached to the Olympus fluorescent microscope (Hachioji-shi, Tokyo, Japan). The micrography of cell morphology and LY diffusion into cellular monolayer after scrape-loading were shown (upper panel: magnification, ×200; lower panel: magnification, ×400). Note that there was no interruption in LY diffusion between morphologically normal and abnormal cells. No clear boundary was observed (arrow).
Figure S2. Effect of hygromycin on Cx43 protein levels. NRK-E52 cells were exposed to various concentrations of hygromycin for 24 h. Cellular proteins were extracted and subjected to Western blot analysis using an anti-Cx43 antibody NP and P denote nonphosphorylated and phosphorylated Cx43 respectively. Equal loading of protein per lane was verified by reprobing the blots with an anti-β-actin antibody. Note the obvious shift of Cx43 from NP to P after hygromycin treatment.
Figure S3. Effect of α-glycyrrhetinic acid (α -GA) on G418-elicited cytotoxicity in NRK cells, as revealed by Dead Red (Ethidium Homodimer-2) Staining. NRK cells in 96-well plates were exposed to 1000 μg·mL−1 G418 for 48 h. The cell viability was assessed by a Cell Viability Assay Kit from Invitrogen following the manufacturer's instruction (LIVE/DEAD Viability/Cytotoxicity Kit, Invitrogen, Carlsbad, CA, USA). The red fluorescent cells represent the dead cells. Note the obvious reduced number of dead cells in the presence of 5 μM α -GA. Magnification, ×200.
Figure S4. Effect of various gap junction inhibitors on G418-induced cell toxicity in NRK-E52 cells. NRK-E52 cells in 12-well plates were exposed to the indicated concentration of G418 in the presence or absence of 5 μM α-glycyrrhetinic acid (α -GA), 3 mM heptanol or 100 μM lindane for 48 h. The detached cells were washed out and the adherent cells were photographed. Note the obvious more attached cells in the presence of gap junction inhibitors. Magnification, ×200.
Figure S5. Forced expression of mutant Cx43 in LLC-PK1 cells does not affect cell susceptibility to hygromycin. LLC-PK1 cells were transfected with a vector encoding GFP protein (pEGFP), wild-type Cx43 (Cx43-EGFP) or mutant Cx43 (Δ130–137 Cx43 by Oyamada et al., 2002) and clones expressing a high level of GFP or Cx43 were selected and analysed for their responses to hygromycin. Cells were treated with the indicated concentrations of hygromycin for 48 h. Cell viability was evaluated by WST assay. Data are expressed as percentage of living cells against the untreated control. Note that forced expression of the mutant Cx43 in LLC-PK1 cells did not sensitize them to hygromycin.
Figure S6. Close correlation between Cx43 protein level and cell susceptibility to G418. (A) Cx43 protein levels in fetal forearm fibroblasts derived from Cx43 wild-type (Cx43+/+), heterozygous (Cx43+/–) and knockout (Cx43–/–) littermates. Cellular protein was extracted from primarily cultured fetal fibroblasts and subjected to Western blot analysis of Cx43. Note the different levels of Cx43 in fibroblasts from Cx43+/+, +/– and –/– littermates. (B) Gap junctional intercellular communication (GJIC) in fetal fibroblasts. Fibroblasts were confluently cultured. A scrape line on the monolayer of fibroblasts was made with a surgical blade. Lucifer Yellow (LY) was added and allowed for diffusion for 3 min. After washing, the cells were fixed and photographed with a digital camera attached to the Olympus fluorescent microscope (Hachioji-shi, Tokyo, Japan). Note the different ability in GJIC among Cx43+/+, +/– and –/– fibroblasts. (C) G418-elicited cell injury in fetal fibroblasts expressing different amounts of Cx43. Fibroblasts were exposed to the indicated concentrations of G418 for 48 h. Cell viability was evaluated by WST assay. The data were expressed as percent of control (mean + s.e., n = 4). *P < 0.01. Note that the degree of cell injury induced by G418 was closely correlated with the levels of Cx43, being severe in Cx43+/+ cells, less severe in Cx43+/– cells and the least severe in Cx43–/– cells.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam EI, de Toledo SM, Little JB. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha -particle irradiated to nonirradiated cells. PNAS. 2001;98:473–478. doi: 10.1073/pnas.011417098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WH, Pak K, Lin B, Webster N, Ryan AF. A PI3K pathway mediates hair cell survival and opposes gentamicin toxicity in neonatal rat organ of Corti. J Assoc Res Otolaryngol. 2006;7:373–382. doi: 10.1007/s10162-006-0050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronier L, Frendo JL, Defamie N, Pidoux G, Bertin G, Guibourdenche J, et al. Requirement of gap junctional intercellular communication for human villous trophoblast differentiation. Biol Reprod. 2003;69:1472–1480. doi: 10.1095/biolreprod.103.016360. [DOI] [PubMed] [Google Scholar]
- Dilber MS, Abedi MR, Christensson B, Bjorkstrand B, Kidder GM, Naus CC, et al. Gap junctions promote the bystander effect of herpes simplex virus thymidine kinase in vivo. Cancer Res. 1997;57:1523–1528. [PubMed] [Google Scholar]
- Fillastre JP, Hemet J, Tulkens P, Morin JP, Viotte G, Olier B, et al. Comparative nephrotoxicity of four aminoglycosides: biochemical and ultrastructural modifications of lysosomes. Adv Nephrol Necker Hosp. 1983;12:253–275. [PubMed] [Google Scholar]
- Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res. 2004;61:386–401. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- Guan X, Ruch RJ. Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial cells treated with DDT and lindane. Carcinogenesis. 1996;17:1791–1798. doi: 10.1093/carcin/17.9.1791. [DOI] [PubMed] [Google Scholar]
- Guo Y, Martinez-Williams C, Gilbert KA, Rannels DE. Inhibition of gap junction communication in alveolar epithelial cells by 18alpha-glycyrrhetinic acid. Am J Physiol. 1999;276:L1018–L1026. doi: 10.1152/ajplung.1999.276.6.L1018. Pt 1. [DOI] [PubMed] [Google Scholar]
- Haefliger JA, Demotz S, Braissant O, Suter E, Waeber B, Nicod P, et al. Connexins 40 and 43 are differentially regulated within the kidneys of rats with renovascular hypertension. Kidney Int. 2001;60:190–201. doi: 10.1046/j.1523-1755.2001.00786.x. [DOI] [PubMed] [Google Scholar]
- Hillis GS, Duthie LA, Mlynski R, McKay NG, Mistry S, MacLeod AM, et al. The expression of connexin 43 in human kidney and cultured renal cells. Nephron. 1997;75:458–463. doi: 10.1159/000189585. [DOI] [PubMed] [Google Scholar]
- Huang RP, Hossain MZ, Huang R, Gano J, Fan Y, Boynton AL. Connexin 43 (cx43) enhances chemotherapy-induced apoptosis in human glioblastoma cells. Int J Cancer. 2001;92:130–138. [PubMed] [Google Scholar]
- Imanaga I, Hai L, Ogawa K, Matsumura K, Mayama T. Phosphorylation of connexin in functional regulation of the cardiac gap junction. Exp Clin Cardiol. 2004;9:161–164. [PMC free article] [PubMed] [Google Scholar]
- Jin QH, Zhao B, Zhang XJ. Cytochrome c release and endoplasmic reticulum stress are involved in caspase-dependent apoptosis induced by G418. Cell Mol Life Sci. 2004;61:1816–1825. doi: 10.1007/s00018-004-4143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovskikh VA, Piccoli C, Yamasaki H. Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene. 2002;21:1989–1999. doi: 10.1038/sj.onc.1205187. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Mussche S, Leybaert L, D'Herde K. Gap junctional communication and connexin43 expression in relation to apoptotic cell death and survival of granulosa cells. J Histochem Cytochem. 2004;52:1199–1207. doi: 10.1369/jhc.3A6227.2004. [DOI] [PubMed] [Google Scholar]
- Krystal GW, Sulanke G, Litz J. Inhibition of phosphatidylinositol 3-kinase-Akt signaling blocks growth, promotes apoptosis, and enhances sensitivity of small cell lung cancer cells to chemotherapy. Mol Cancer Ther. 2002;1:913–922. [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Kuwana H, Terada Y, Kobayashi T, Okado T, Penninger JM, Irie-Sasaki J, et al. The phosphoinositide-3 kinase gamma-Akt pathway mediates renal tubular injury in cisplatin nephrotoxicity. Kidney Int. 2008;73:430–445. doi: 10.1038/sj.ki.5002702. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–3442. doi: 10.1242/jcs.03089. Pt 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AF. c-Src: bridging the gap between phosphorylation- and acidification-induced gap junction channel closure. Sci STKE. 2005;2005:pe33. doi: 10.1126/stke.2912005pe33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, et al. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Marconi P, Tamura M, Moriuchi S, Krisky DM, Niranjan A, Goins WF, et al. Connexin 43-enhanced suicide gene therapy using herpesviral vectors. Mol Ther. 2000;1:71–81. doi: 10.1006/mthe.1999.0008. [DOI] [PubMed] [Google Scholar]
- Meier M, Nitschke M, Hocke C, Kramer J, Jabs W, Steinhoff J, et al. Insulin inhibits caspase-3 activity in human renal tubular epithelial cells via the PI3-kinase/Akt pathway. Cell Physiol Biochem. 2008;21:279–286. doi: 10.1159/000129386. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003;34:1987–1993. doi: 10.1161/01.STR.0000079814.72027.34. [DOI] [PubMed] [Google Scholar]
- Nakase T, Sohl G, Theis M, Willecke K, Naus CC. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–2075. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyamada Y, Zhou W, Oyamada H, Takamatsu T, Oyamada M. Dominant-negative connexin43-EGFP inhibits calcium-transient synchronization of primary neonatal rat cardiomyocytes. Exp Cell Res. 2002;273:85–94. doi: 10.1006/excr.2001.5411. [DOI] [PubMed] [Google Scholar]
- Ozog MA, Siushansian R, Naus CC. Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. J Neuropathol Exp Neurol. 2002;61:132–141. doi: 10.1093/jnen/61.2.132. [DOI] [PubMed] [Google Scholar]
- Pahujaa M, Anikin M, Goldberg GS. Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp Cell Res. 2007;313:4083–4090. doi: 10.1016/j.yexcr.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Parthasarathi K, Ichimura H, Monma E, Lindert J, Quadri S, Issekutz A, et al. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Invest. 2006;116:2193–2200. doi: 10.1172/JCI26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procida K, Jorgensen L, Schmitt N, Delmar M, Taffet SM, Holstein-Rathlou NH, et al. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm. 2009;6:1632–1638. doi: 10.1016/j.hrthm.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Shao Q, Igdoura SA, Alaoui-Jamali MA, Laird DW. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J Biol Chem. 2003;278:30005–30014. doi: 10.1074/jbc.M300614200. [DOI] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Sato H, Senba H, Virgona N, Fukumoto K, Ishida T, Hagiwara H, et al. Connexin 32 potentiates vinblastine-induced cytotoxicity in renal cell carcinoma cells. Mol Carcinog. 2007;46:215–224. doi: 10.1002/mc.20267. [DOI] [PubMed] [Google Scholar]
- Sha SH, Schacht J. Stimulation of free radical formation by aminoglycoside antibiotics. Hear Res. 1999;128:112–118. doi: 10.1016/s0378-5955(98)00200-7. [DOI] [PubMed] [Google Scholar]
- Shintani-Ishida K, Unuma K, Yoshida K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ J. 2009;73:1661–1668. doi: 10.1253/circj.cj-09-0079. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takens-Kwak BR, Jongsma HJ, Rook MB, Van Ginneken AC. Mechanism of heptanol-induced uncoupling of cardiac gap junctions: a perforated patch-clamp study. Am J Physiol. 1992;262:C1531–C1538. doi: 10.1152/ajpcell.1992.262.6.C1531. Pt 1. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Yao J, Morioka T, Oite T. PDGF regulates gap junction communication and connexin43 phosphorylation by PI 3-kinase in mesangial cells. Kidney Int. 2000;57:1915–1926. doi: 10.1046/j.1523-1755.2000.00041.x. [DOI] [PubMed] [Google Scholar]
- Yao J, Morioka T, Li B, Oite T. Coordination of mesangial cell contraction by gap junction - mediated intercellular Ca(2+) wave. J Am Soc Nephrol. 2002;13:2018–2026. doi: 10.1097/01.asn.0000023436.71816.56. [DOI] [PubMed] [Google Scholar]
- Yao J, Hiramatsu N, Zhu Y, Morioka T, Takeda M, Oite T, et al. Nitric oxide-mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J Am Soc Nephrol. 2005;16:58–67. doi: 10.1681/ASN.2004060453. [DOI] [PubMed] [Google Scholar]
- Yao J, Kitamura M, Zhu Y, Meng Y, Kasai A, Hiramatsu N, et al. Synergistic effects of PDGF-BB and cAMP-elevating agents on expression of connexin43 in mesangial cells. Am J Physiol. 2006;290:F1083–F1093. doi: 10.1152/ajprenal.00134.2005. [DOI] [PubMed] [Google Scholar]
- Yao J, Zhu Y, Morioka T, Oite T, Kitamura M. Pathophysiological roles of gap junction in glomerular mesangial cells. J Membr Biol. 2007a;217:123–130. doi: 10.1007/s00232-007-9023-2. [DOI] [PubMed] [Google Scholar]
- Yao J, Zhu Y, Sun W, Sawada N, Hiramatsu N, Takeda M, et al. Irsogladine maleate potentiates the effects of nitric oxide on activation of cAMP signalling pathways and suppression of mesangial cell mitogenesis. Br J Pharmacol. 2007b;151:457–466. doi: 10.1038/sj.bjp.0707255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Oite T, Kitamura M. Gap junctional intercellular communication in the juxtaglomerular apparatus. Am J Physiol. 2009;296:F939–F946. doi: 10.1152/ajprenal.90612.2008. [DOI] [PubMed] [Google Scholar]
- Yaoita E, Yao J, Yoshida Y, Morioka T, Nameta M, Takata T, et al. Up-regulation of connexin43 in glomerular podocytes in response to injury. Am J Physiol. 2002;161:1597–1606. doi: 10.1016/s0002-9440(10)64438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Kaneda M, Morita I. The gap junction-independent tumor-suppressing effect of connexin 43. J Biol Chem. 2003;278:44852–44856. doi: 10.1074/jbc.M305072200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.