

Abstract
BACKGROUND AND PURPOSE
Efavirenz (EFV) is widely used in the treatment of HIV-1 infection. Though highly efficient, there is growing concern about EFV-related side effects, the molecular basis of which remains elusive.
EXPERIMENTAL APPROACH
In vitro studies were performed to address the effect of clinically relevant concentrations of EFV (10, 25 and 50 µM) on human hepatic cells.
KEY RESULTS
Cellular proliferation and viability were reduced in a concentration-dependent manner. Analyses of the cell cycle and several cell death parameters (chromatin condensation, phosphatidylserine exteriorization, mitochondrial proapoptotic protein translocation and caspase activation) revealed that EFV triggered apoptosis via the intrinsic pathway. In addition, EFV directly affected mitochondrial function in a reversible manner, inducing a decrease in mitochondrial membrane potential and an increase in mitochondrial superoxide production, followed by a reduction in cellular glutathione content. The rapidity of these actions rules out any involvement of mitochondrial DNA replication, which, until now, was thought to be the main mechanism of mitochondrial toxicity of antiretroviral drugs. Importantly, we also observed an increase in mitochondrial mass, manifested as an elevated cardiolipin content and enhanced expression of mitochondrial proteins, which was not paralleled by an increase in the mtDNA/nuclear DNA copy number ratio. The toxic effect of EFV was partially reversed by antioxidant pretreatment, which suggests ROS generation is involved in this effect.
CONCLUSION AND IMPLICATIONS
Clinically relevant concentrations of EFV were shown to be mitotoxic in human hepatic cells in vitro, which may be pertinent to the understanding of the hepatotoxicity associated with this drug.
Keywords: reactive oxygen species, antiretroviral drugs, hepatotoxicity, cell death, side effects, Efavirenz, mitochondria
Introduction
Chronic administration of the various drugs included under the term highly active antiretroviral therapy (HAART) has changed the prognosis of AIDS. Current practice guidelines recommend the combined use of non-nucleoside-reverse-transcriptase inhibitor (NNRTI) or boosted protease inhibitor regimens with two nucleoside-reverse-transcriptase inhibitors (NRTI) (Hammer et al., 2008; AIDSInfo, 2010). Due to the special characteristics of the disease, the development of these drugs was particularly rapid and focused essentially on clinical efficacy, that is, reduction in mortality. However, as the disease has become a controlled condition, there has been increasing emphasis placed on the long-term adverse effects induced by this life-long pharmacological treatment. The mitochondrion is a major target of drug-induced cytotoxicity, which occurs through a wide variety of mechanisms such as inhibition or uncoupling of oxidative phosphorylation, oxidative stress and/or opening of the mitochondrial permeability transition pore (Labbe et al., 2008). Many important adverse effects associated with HAART are known to be the consequence of mitochondrial toxicity, but they have been mainly attributed to the inhibition by NRTI of mitochondrial DNA polymerase-γ (Pol-γ), the enzyme responsible for mtDNA replication (Martin et al., 1994; Walker et al., 2002). The effects on mitochondria of NNRTIs, which do not inhibit Pol-γ, are less well documented, though some elements of the toxicity attributed to these drugs resemble disorders induced by mitochondrial dysfunction (Sato, 2007; Abdul-Ghani and DeFronzo, 2008).
Efavirenz (EFV) is the most widely used NNRTI, and although considered a safe drug, there is growing concern that EFV-containing therapies are associated with lipid and metabolic disorders, psychiatric symptoms and hepatotoxicity (Tashima et al., 2003; Gutierrez et al., 2005; Manfredi et al., 2005). The clinical manifestations of these events have been characterized, but the cellular and molecular mechanisms underlying these detrimental effects of EFV remain largely unknown.
Up to 10% of HIV patients treated with EFV exhibit increases in liver enzymes that may require the treatment to be discontinued (Kappelhoff et al., 2005). In the present study, we show that clinically relevant concentrations of EFV induce a rapid mitotoxic effect in human hepatic cells by a mechanism independent of mitochondrial DNA replication. EFV reduced cellular proliferation and viability in a concentration-dependent manner, triggered apoptosis via the intrinsic pathway and modified several parameters of mitochondrial function, including the induction of oxidative stress and a significant increase in mitochondrial mass. The fact that some of these harmful effects were partially reversed by an antioxidant treatment suggests that ROS generation is implicated in their manifestation.
Methods
Reagents
Unless otherwise stated, chemical reagents were purchased from Sigma-Aldrich (Steinheim, Germany). EFV (Sustiva® 600 mg, Bristol-Myers Squibb, Princeton, NJ, USA) was obtained from its clinically available preparation and, once insoluble substances were removed by filtration, was dissolved in methanol (3 mg·mL−1). The purity (98–100%) and stability of the solutions were evaluated by high-performance liquid chromatography (HPLC) and compared with a control solution of EFV purchased from Sequoia Research Products (Pangbourne, UK). No differences were detected in the stability and the activity between this control solution and EFV obtained from the galenic form. A concentration of 0.5% methanol was employed in all EFV treatments and vehicle control experiments, versus which the statistical analysis was always performed. This concentration of methanol did not have a significant impact on any of the parameters studied.
Cell culture
Experiments were performed with the human hepatoblastoma cell line Hep3B (ATCC HB-8064). In specific cases, primary human hepatocytes obtained from biopsies of patients (three women, three men) undergoing surgical resection of liver tumours (Hospital ‘La Fe’, Valencia, Spain) were employed. Several experiments were also reproduced in HUVEC obtained from fresh human umbilical cords (Department of Gynaecology, Hospital Clínico Universitario, Valencia, Spain) and the human cervical carcinoma cell line HeLa (ATCC CCL-2). Unless stated otherwise, all reagents employed in cell culture were purchased from Gibco (Invitrogen, Eugene, OR, USA). Hep3B cells were cultured in MEM supplemented with 1 mM non-essential amino acids. HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with a high glucose concentration (4.5 mg·mL−1). Both Hep3B and HeLa culture media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (50 U·mL−1) and streptomycin (50 µg·mL−1). HUVEC were isolated by extraction with collagenase (Jaffe et al., 1973) and cultured in EMG-2 medium supplemented with BulletKit components (Clonetics, Lonza, Walkersville, MD, USA) according to the manufacturer's instructions, and with 50 U·mL−1 penicillin, 50 µg·mL−1 streptomycin and 2.5 µg·mL−1 fungizone-Amphotericin B. Hepatocytes were isolated from biopsies following a two-step collagenase protocol (Pichard et al., 2006). In brief, biopsies were first perfused (15 min) with dissociation solution containing 0.05% collagenase, 20 mM HEPES, 120 mM NaCl, 5 mM KCl, 0.7 mM CaCl2, 0.5% glucose, 100 µM sorbitol, 100 µM manitol, 100 µM glutathione (GSH), 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 0.25 µg·mL−1 amphotericin B, pH 7.2. Further homogenization was achieved with the addition of 0.2% collagenase (37°C, 20 min). The resulting cell suspension was then filtered through a nylon mesh (250 µm) and washed three times at 50×g (5 min) in a culture medium containing advanced D-MEM/F12 and William's E medium (1:1) supplemented with 0.29 g·L−1 glutamine, 0.04 mg·L−1 dexamethasone and 200 µg·L−1 glucagon. Cell viability (>80%) was determined by trypan blue exclusion. Hepatocytes were seeded in type I collagen-coated dishes (BD labware, Oxford, UK) and maintained for 72 h in a culture medium containing 5% FBS, that was refreshed every 24 h. A stable cell culture was obtained in which at least 90% of cells were hepatocytes.
All cell cultures were maintained in an incubator (IGO 150, Jouan, Saint-Herblain Cedes, France) at 37°C in a humidified atmosphere of 5% CO2/95% air (AirLiquide Medicinal, Valencia, Spain). Treatments were always performed in complete cell culture medium. The protocols employed complied with European Community guidelines for the use of human experimental models and were approved by the Ethics Committee of the University of Valencia.
Proliferation and viability
Cells were seeded in 6-well plates, allowed to proliferate exponentially for 3 days in the presence of EFV and counted at 24, 48 and 72 h using a haemacytometer (Bright Line Counting Improved Neubauer Chamber, Hausser Scientific, Horsham, PA, USA). In addition, we performed the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diprenyl tetrazolium bromide] assay, which is a colorimetric assay based on the ability of cells to reduce a soluble yellow tetrazolium salt to blue formazan crystals (Mosmann, 1983). This reduction takes place only when mitochondrial reductase enzymes are active, and is thus a marker of cell viability related to mitochondrial function. EFV treatment was performed over a 24 h-period, in 96-well plates. MTT reagent (Roche Diagnostics, Mannheim, Germany) was added (20 µL per well) for the last 4 h of the treatment. Cells were dissolved in DMSO (100 µL per well, 5 min, 37°C) and absorbance was measured using a ‘Multiscan’ plate-reader spectrophotometer (Thermo Labsystems, Thermo Scientific, Rockford, IL, USA).
Protein extracts and Western blotting
Protein extracts were obtained using t-75 flask cell cultures of Hep3B cells. To obtain mitochondria-enriched protein fraction, cells were lysed with 0.5 mL fractionation buffer (10 mM Tris-HCL pH 7.5, 0.25 M sucrose and 1 mM EDTA) on ice by passage through a 23-gauge needle. Unbroken cells were pelleted by 10 min centrifugation at 500×g at 4°C. The lysate was centrifuged again at 16100×g for 30 min at 4°C. The pellet representing the mitochondrial fraction was suspended in 50 µL mitochondrial buffer (10 mM Tris-acetate pH 8.0, 0.5% NP-40, 5 mM CaCl2, 1 mM DTT and ‘Complete Mini’ protease inhibitor cocktail from Roche Diagnostics). Whole-cell protein extracts were obtained by lysing cell pellets in 50–100 µL complete lysis buffer (20 mM HEPES pH 7.4, 400 mM NaCl, 20% (v/v) glycerol, 0.1 mM EDTA, 10 µM Na2MoO4, 1 mM DTT) supplemented with protease inhibitors (‘Complete Mini’ protease inhibitor cocktail, and ‘Pefabloc’, both from Roche Diagnostics), and phosphatase inhibitors mixture: 10 µM NaF, 10 mM NaVO3, 10 mM p-nitrophenylphosphate (PNPP) and 10 mM β-glycerolphosphate. Samples were vortexed, incubated on ice for 15 min, vortexed again and centrifuged in a microcentrifuge at 16100×g for 15 min at 4°C. Protein content was quantified employing the ‘BCA Protein Assay Kit’ (Pierce, Thermo Scientific). SDS-PAGE and WB were performed using standard methods (Bio-Rad, Hercules, CA, USA), with 40–50 µg of the protein extract, and employing primary antibodies: anti-AIF (ψProSci, Poway, CA, USA) at 1:500 dilution, anti-actin at 1:500 (Sigma-Aldrich), anti-cyt c at 1:1000 (Zymed, Invitrogen, Carlsbad, CA, USA), anti-Complex I subunits 17, 20 and 39 kDa, anti-Complex IV subunit II, anti-Complex V subunits α and β and anti-Porin, all at 1:1000 and from Molecular Probes (Invitrogen) and secondary antibodies: peroxidase-labelled anti-rabbit IgG from Vector laboratories (Burlingame, CA, USA) at 1:5000 and anti-mouse antibody from Dako (Glostrup, Denmark) at 1:2000. Immunolabelling was detected using the enhanced chemiluminescent reagent ECL (Amersham, GE Healthcare, Little Chalfont, UK) or SuperSignal WestFemto (Pierce, Thermo Scientific), and was visualized with a digital luminescent image analyser (FUJIFILM LAS 3000, Fujifilm). ImageQuant software version 4.0. was used for densitometric analysis.
Fluorescence microscopy and static cytometry
All treatments were performed in duplicate in 24-well plates, and 16–30 images per well were recorded with a fluorescence microscope (IX81, Olympus, Hamburg, Germany) coupled with a static cytometry software ‘ScanR’ version 2.03.2 (Olympus).
Apoptosis
Bivariate Annexin V/PI analysis
Following 24 h treatment with EFV, the medium was replaced with HBSS containing 1 µM of the chromatin-specific dye Hoechst 33342 and 0.5 µL AnnexinV-Fluorescein in order to detect phosphatidylserine exteriorization, and was incubated in the dark for 15 min at 37°C. Thereafter, 0.5 µL of the chromatin-detecting dye propidium iodide (PI) was added (5 min) to label dead or damaged cells. Annexin V and PI solutions were purchased in the form of Annexin-V-FLUOS Staining kit (Roche Diagnostics). Staurosporine (STS), a widely used protein kinase inhibitor, was employed as a positive pro-apoptotic control (Lakhani et al., 2006).
Detection of caspase-3, -9 and caspase-8
Activated caspases can be detected in living cells with specific cell-permeable, non-toxic fluorescent dyes. We used CaspGLOW™– Red Active Caspase-9, – Fluorescein Active Caspase-3 and – Red Active Caspase-8 Staining kits (Biovision Research Products, Mountain View, CA, USA). The assay employs RED-LEHD-FMK (caspase-9), fluorescein isothiocyanate (FITC)-DEVD-FMK (caspase-3) and RED-IETD-FMK (caspase-8) as fluorescent markers. Cells were treated with EFV for 24 h using 1 µM STS as a positive control. The fluorochromes for caspase detection and 1 µM Hoechst 33342 were subsequently added and incubated in the dark for 30 min (37°C).
Mitochondrial membrane potential and superoxide detection
Cells were treated with EFV or the vehicle for a period of 1, 6 and 24 h. Fluorochromes 1 µM Hoechst 33342 and 5 µM TMRM (to detect membrane potential) or 5 µM MitoSOX (to detect mitochondrial superoxide), both from Molecular Probes (Invitrogen), were added for the last 30 min of treatment. FCCP (uncoupler of oxidative phosphorylation) or Rotenone (Complex I inhibitor) was used as a positive control (Labbe et al., 2008). To assess the reversibility of the effects, cells were treated for 1 h with EFV, which was subsequently removed. Then, 30 min later, the fluorochromes were added for a further 30 min incubation. The fluorescence detection filter set for both TMRM and MitoSOX was excitation max 540/10 nm, dichroic filter 570 nm and emission 590 nm LP.
The MitoSOX signal was also detected using a ‘Fluoroscan’ multi-well plate reader (Thermo Labsystems, Thermo Scientific), excitation/emission maxima of 390/530 nm. Hep3B cells were incubated with EFV for 24 h in 96-well plates and 5 µM MitoSOX was added for the last 30 min. Cells were then washed with HBSS and fluorescence was immediately recorded. Fluoresence was also evaluated prior to addition of MitoSOX, as autofluorescence can be intense at near-UV wavelength.
Determination of mitochondrial mass
The fluorescent dye 10-N-nonyl-acridine orange chloride (NAO) (Sigma-Aldrich), which specifically binds to cardiolipin independently of the mitochondrial membrane potential, was used to monitor mitochondrial mass (Wu et al., 2007). Cells were treated with EFV or vehicle for 1, 6 or 24 h. Fluorochromes (1 µM Hoechst 33342 and 0.5 µM NAO) were added for the last 30 min of the treatment and incubated in the dark. These experiments were also performed using the fluorescent mitochondrial stain Mitotracker Green FM 5 µM (Molecular Probes, Invitrogen).
In order to study the involvement of ROS in the effect of EFV, several static cytometry experiments were performed where the cells were treated with Trolox, a water-soluble analogue of vitamin E (Forrest et al., 1994). In these experiments, cells were cultured in the presence of Trolox for 1 h and then EFV was added for a further 24 h incubation, without the medium being refreshed.
Detection of GSH
GSH was detected by a fluorometric method using monochlorobimane (Molecular Probes, Invitrogen), which crosses the plasma membrane and forms an intracellular fluorescent adduct in a reaction catalysed by GSH S-transferase (Sebastiáet al., 2003). Following 4 h treatment with EFV in 96-well plates, the culture medium was discarded and 100 µL HBSS containing 0.04 mM monochlorobimane were added to each well, which was followed by a 30 min incubation in the dark, at 37°C. Fluorescence was detected using a Fluoroscan (Thermo Labsystems, Thermo Scientific) at λexc of 355 nm and λemis of 460 nm.
Determination of mtDNA copy number
Total cellular DNA was isolated using the QIAamp® DNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions and quantified using the NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). DNA purity was verified by standard electrophoresis using agarose gels. For the amplification of mtDNA content, we used the following primers, which are complementary to sequences of the NDR1 gene: mtF3212 5′-CACCCAAGAACAGGGTTTGT-3′ (forward) and mtR3319 5′-TGGCCATGGGTATGTTGTTTA-3′ (reverse). mtDNA content was quantified relative to nuclear DNA (nDNA), for the amplification of which we used primers complementary to sequences of the nuclear gene cyclophylin A: CypAF 5′CGTCTCCTTTGAGCTGTTTG-3′ (forward) and CypAR 5'TCTGGTCGTTCTTCTAGTGG-3′ (reverse). PCR reactions were performed in a Carousel-based LightCycler® 2.0 Real Time PCR System (Roche Applied Biosystems). Both PCR reactions of mtDNA and nDNA were performed using 25 ng total DNA, mixed with LightCycler® FastStart DNA MasterPLUS SYBR Green I master mix (Roche Applied Science) following instructions in the manual. Forward and reverse primers (0.5 µM for NDR1 and 1 µM for CYPA) were added in a final reaction volume of 10 µL. For mtDNA amplification, reactions were performed as follows: 5 min denaturalization at 95°C, followed by 40 cycles of 1 s at 95°C, 5 s at 65°C and 6 s at 72°C. For nuclear DNA amplification, reactions were performed as follows: 10 min denaturalization at 95°C, followed by 35 cycles of 1 s at 95°C, 5 s at 58°C and 18 s at 72°C. Standard curve was included in each run and the specificity of the amplified products was verified by melting curve analysis and agarose gel electrophoresis. Data were calculated as number of DNA copies. The results shown are a summary of 3 independent experiments (in duplicate) and are expressed as a ratio of mtDNA/nDNA copy.
Presentation of data and statistical analysis
Data were analysed using GraphPad Prism v.3. software with one-way anova, followed by Newman–Keuls multiple comparison test or Student's t-test. All values are expressed as mean ± SEM and the results were considered to be statistically significant when: *P < 0.05, **P < 0.01 and ***P < 0.001. In most cases data are represented as percent of control, the negative control (untreated cells) being considered to be 100%.
Results
EFV treatment reduced cellular proliferation and viability
Figure 1A shows the effect of EFV on cell survival and proliferation of Hep3B cells. During the 3 day experimental period evaluated, no significant differences in growth rate were detected in cells treated with 10 µM. However, EFV 25 µM inhibited cell growth, as the number of cells counted was lower than in the control at 24 h and remained significantly lower throughout the 72 h period of culture. EFV 50 µM appeared to be cytotoxic, as most cells did not survive the treatment. The decrease in the number of EFV 50 µM-treated cells was substantial after 24 h and was highly significant following 48 and 72 h (94.61 ± 1.2% and 97 ± 1.12% reduction respectively). Figure 1B shows representative inverted light microscopy images of Hep3B cells after 24 h treatment with EFV at the three concentrations evaluated. It is noteworthy that, in addition to a diminished proliferation, cells exposed to EFV 50 µM displayed clear morphological modifications indicative of cellular stress. The MTT assay, the results of which are shown in Figure 1C, confirmed the inhibitory effect of 24 h treatment with EFV 50 µM and suggested that it was related to a diminished mitochondrial function. In order to determine whether this inhibitory effect of EFV was cell-specific, we also studied cellular proliferation and viability in two other cell types (HeLa and HUVEC) and found the inhibition more severe, reaching statistical significance in both cell lines within the first 24 h of treatment with EFV 25 µM. The absorbance recorded in the MTT assay for HeLa cells was 79.09 ± 7.79% in EFV 25 µM, and 21.56 ± 5.31% in EFV 50 µM (n= 9; *P < 0.05 for EFV 25 µM and ***P < 0.001 for EFV 50 µM vs. control), and the corresponding values for HUVEC were 39.04 ± 11.28% and 3.24 ± 0.29% (n= 6; **P < 0.01 for EFV 25 µM and ***P < 0.001 for EFV 50 µM vs. control).
Figure 1.
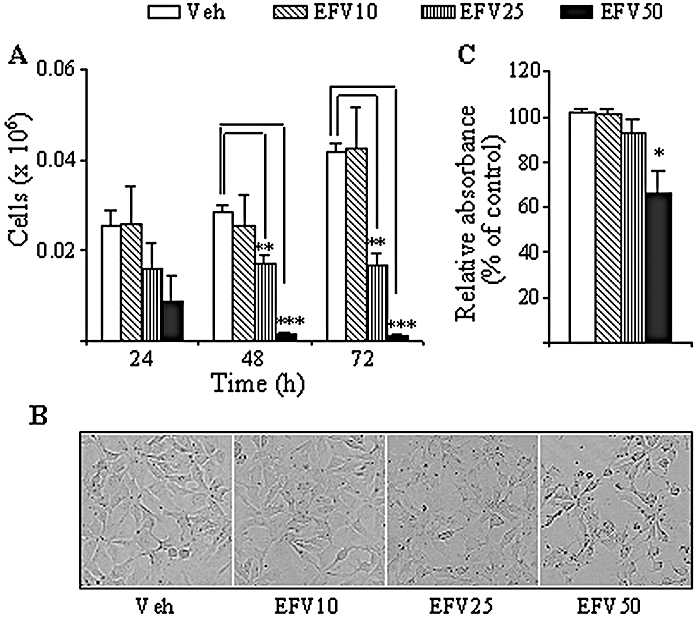
Effect of Efavirenz (EFV) treatment on cellular proliferation and viability of Hep3B cells (A) Cell count over 3 days using a haemacytometer revealed a concentration-dependent inhibition of cellular proliferation. **P < 0.01, ***P < 0.001 versus vehicle. (B) Representative inverted light microscopy images (10x) of cell culture treated with EFV for 24 h. (C) MTT assay of exponentially growing cells after 24 h of culture in the presence of EFV. Data (mean ± SEM, n= 3–6) were analysed by a one-way anova multiple comparison test followed by Newman–Keuls test, significance versus vehicle *P < 0.05.
EFV treatment induced apoptosis
Although a clear trend towards apoptotic changes was observed with 10 and 25 µM EFV, only those induced by 50 µM EFV reached statistical significance after the 24 h incubation period. Figure 2A shows the results of the bivariate Annexin V/PI analysis of exponentially growing Hep3B cells, which was performed by static cytometry. Four different cellular subpopulations were evaluated: vital (double negative), apoptotic (Annexin V+/PI-), late apoptotic/necrotic (Annexin V+/PI+) and damaged cells (Annexin V-/PI+) cells. The cytogram shows a representative experiment in which EFV 50 µM-treated cells displayed apoptotic features: 9.3% of cells were Annexin V+/PI- compared with only 0.5% of the negative control counterparts and 29.2% of STS 1 µM-treated cells (positive control). Figure 2B shows representative fluorescence microscopy images of Hoechst-stained nuclei. Hep3B cells undergoing treatment with 50 µM EFV exhibited typical apoptotic features, such as chromatin condensation and nuclear fragmentation. We also quantified the mean intensity of Hoechst fluorescence in each nucleus and observed a consistent increase, paralleled by a decrease in the nuclear area of Hoechst fluorescence, both hallmarks of chromatin condensation. Although a trend was observed, the changes in the bivariate Annexin V/PI analysis and the intensity of Hoechst fluorescence in cells treated with lower concentrations of EFV (10 and 25 µM) were not statistically significant. Conversely, the mean area of Hoechst fluorescence in cells treated with EFV 10 µM was 99.52 ± 0.55%, this value decreased to 97.71 ± 1.39% and 94.57 ± 1.11%, in those treated with 25 µM and 50 µM EFV, respectively, whereas the mean Hoechst fluorescence augmented gradually from 103.23 ± 1.56% in cells treated with EFV10 µM, to 105.97 ± 1.5% and 123.82 ± 3.33% with EFV 25 and 50 µM respectively (all values compared with the control value of 100%). We also analysed the expression of cyt c and AIF, two pro-apoptotic mitochondrial proteins which translocate to the cytosol and the nucleus, respectively, during apoptosis (Danial and Korsmeyer, 2004; Li and Yuan, 2008). Incubation with EFV for 24 h produced a concentration-dependent reduction in the expression of both proteins in mitochondria-enriched cellular fractions, which reached statistical significance only with EFV 50 µM (Figure 2C). The presence of apoptotic cells was also observed during cell cycle analysis. Figure 3A and B shows a significant increase in the subG1 peak in EFV 50 µM-treated cells with respect to control. No significant changes were observed with the lower concentrations of EFV employed. Of note, EFV 50 µM treatment also displayed a significantly lower percentage of cells in the G1 peak and an increase in the G2M subpopulation, pointing to a cell cycle arrest in G2M. It should be pointed out that the static cytometry experiments confirmed the cytotoxic effect of 50 µM EFV on cellular proliferation and viability described in Figure 1 with other techniques. For example, Hoechst fluorescence of 30 fields per well (Figure 2A) detected 8022 cells in the untreated control, 3118 cells with the EFV 50 µM-treatment and 1576 cells with the 1 µM STS-treatment. Further confirmation of the pro-apototic effect of EFV is provided by studying the activation of caspases as one of the early hallmarks of several apoptotic pathways (Danial and Korsmeyer, 2004; Lakhani et al., 2006; Li and Yuan, 2008). Figure 3C shows how EFV 50 µM induced activation of caspase-3 and -9, but not of caspase-8 (data not shown), a finding which points to the triggering of the intrinsic (mitochondrial) but not the extrinsic pathway of apoptosis. A summary showing the cellular population positive for caspase-3 and caspase-9 after 24 h EFV treatment is given in Figure 3D. These experiments revealed that 19.44 ± 8.50% of EFV 50 µM-treated cells were caspase-3 positive whereas 28.55 ± 7.41% proved caspase-9 positive. Interestingly, EFV 25 µM-treated cells only showed caspase-9 activation.
Figure 2.
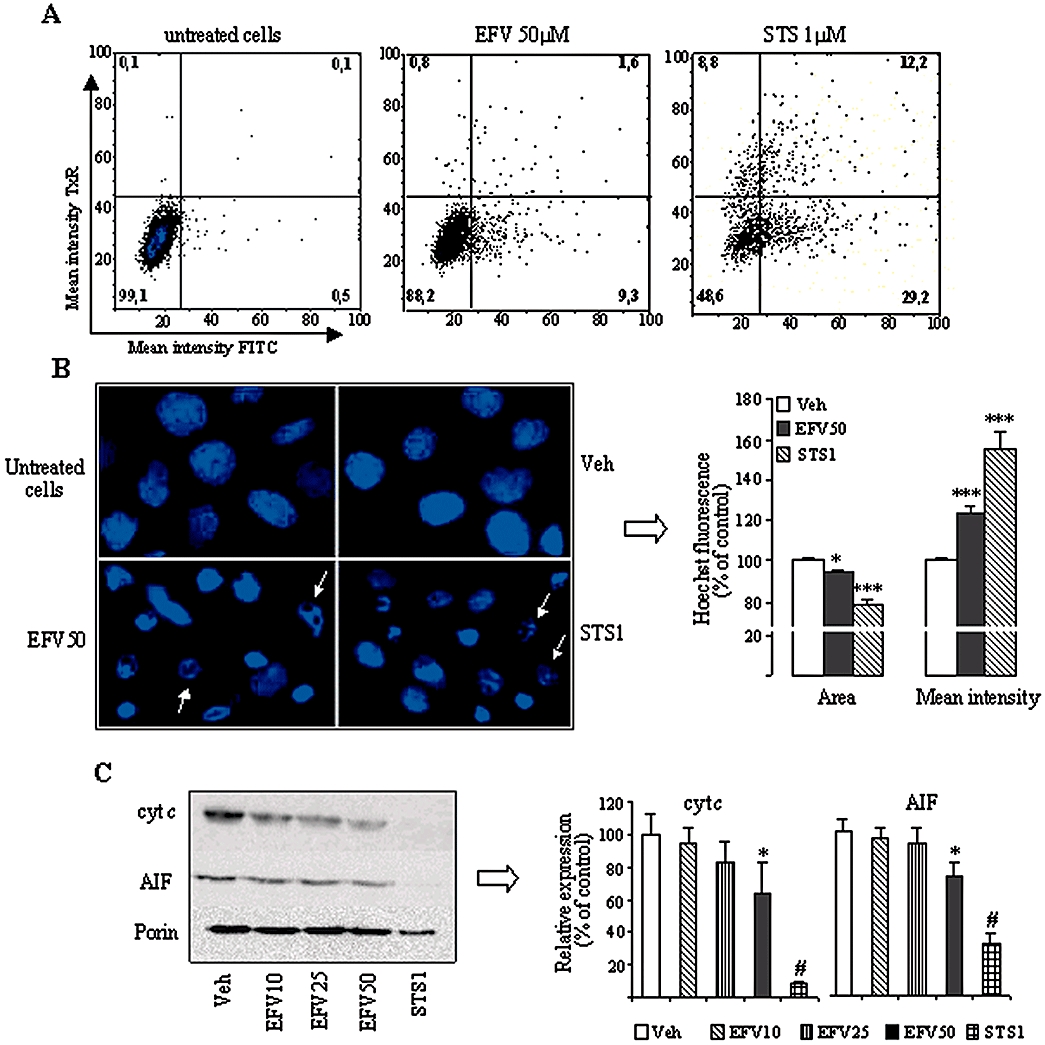
Activation of apoptosis in Hep3B cells after 24 h treatment with Efavirenz (EFV). (A) Representative histograms (Bivariate Annexin V/PI analysis) of untreated control cells, and cells treated with 50 µM EFV and 1 µM STS, showing the existence of 4 cellular populations: AnnV-/PI-, AnnV+/PI-, AnnV-/PI+ and AnnV+/PI+ (% of each population for all three conditions is provided) (B) Nuclear morphology by Hoechst staining. Left panel: representative fluorescence microscopy images (40x). Arrows show typically apoptotic nuclei. Right panel: summary of the Hoechst fluorescence data, calculated as % of control (untreated cells) fluorescence, which was considered 100%. (C) Western blot analysis using mitochondria-enriched cellular fractions. Representative WB image and histogram expressing quantification of protein expression. Quantification was performed by densitometry and the results (mean ± SEM, n= 4–9) are expressed as protein expression in relation to that of untreated cells in each individual experiment, which was considered 100%. Statistical analysis was performed by one-way anova multiple comparison test followed by Newman–Keuls test (*P < 0.05, ***P < 0.001 vs. vehicle). The positive control was independently analysed by Student's t-test (# P < 0.001).
Figure 3.
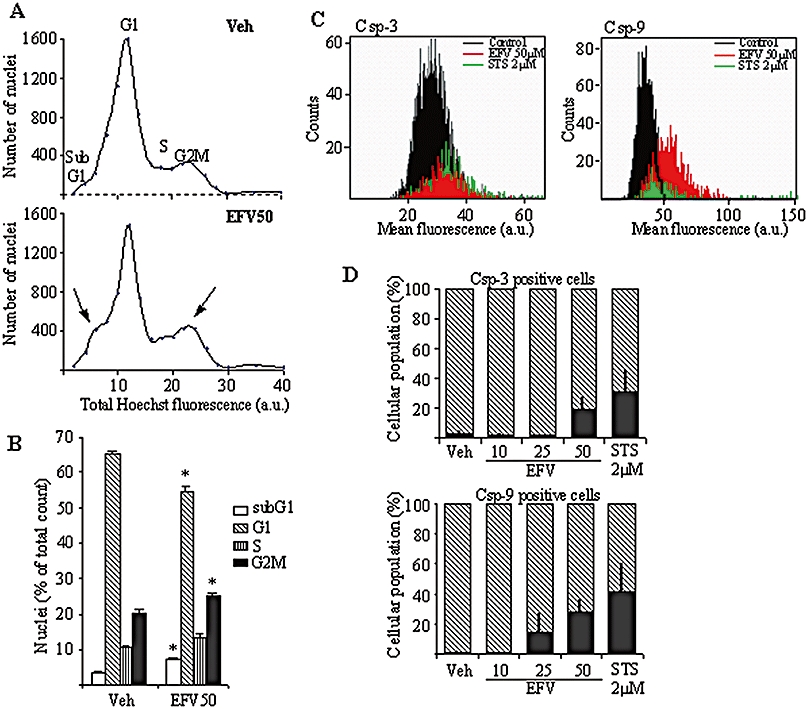
Cell cycle analysis by static cytometry (Hoechst fluorescence) in Hep3B cells treated with vehicle or Efavirenz (EFV) 50 µM for 24 h. Representative cytograms (A) and a summary histogram (B) are shown; statistical analysis was performed by one-way anova multiple comparison test followed by Newman–Keuls test (*P < 0.05 vs. vehicle). (C,D) Analysis of caspase-3 and -9 activation by static cytometry in Hep3B cells treated for 24 h with increasing concentrations of EFV, using 2 µM STS treatment as a control. Representative cytograms (C) show caspase-3 and caspase-9 activation detected as mean fluorescence. The histograms (D) summarize the fluorescence data and represent the % of positive cells in the total cellular population (shaded area). Data shown as mean ± SEM (n= 3–6).
EFV treatment compromised mitochondrial function
To analyse the mitotoxic potential of EFV, we assessed its effect on mitochondrial superoxide production using the mitochondria-targeted fluorochrome MitoSOX. In all three periods evaluated, 1, 6 and 24 h (Figure 4A), incubation of Hep3B cells with EFV 50 µM led to a significant increase in mitochondrial superoxide production, detected as increase in MitoSOX fluorescence. Importantly, the increase in mitochondrial superoxide accumulated over time. In particular, at 6 and 24 h treatment the values obtained with EFV 50 µM were very similar to those obtained with Rotenone, a well-known inducer of superoxide production at Complex I of the mitochondrial electron chain. Unlike in most of the previous experiments with apoptosis, a clear and statistically significant effect was also observed with EFV 25 µM. EFV 10 µM induced no effects. Given that in live cells the selectivity of ethidium-based probes such as MitoSOX for superoxide is complicated by auto-oxidation and superoxide-independent oxidation processes, we performed additional experiments to ensure the specificity of the signal detected. Incubation of Hep3B cells with 50 µM FeTCPP, a SOD mimetic (Liu and Schubert, 2009), largely prevented the EFV-induced increase in MitoSOX fluorescence (data not shown), suggesting that superoxide was responsible for the increase in the fluorescent signal. Moreover, taking into consideration recent reports that the superoxide-dependent MitoSOX oxidation product exhibits higher selectivity with excitation at 390 nm (Robinson et al., 2006), we detected its fluorescence in a multi-well plate fluorometer with excitation/emission maxima at 390/530 nm and confirmed that the increased signal induced by EFV in Hep3B cells after 24 h treatment was concentration-dependent (Supporting Information Figure S1).
Figure 4.
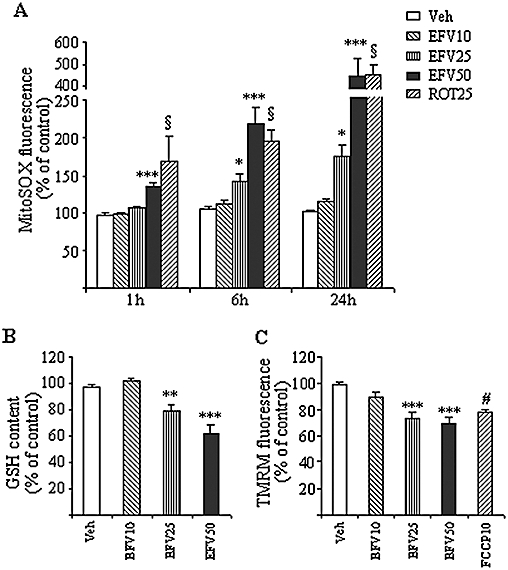
Effect of Efavirenz (EFV) treatment on mitochondrial redox state. (A) Superoxide production. MitoSOX fluorescence in Hep3B cells treated with increasing concentrations of EFV and vehicle for 1, 6 and 24 h using rotenone 25 µM as a positive control. (B) GSH concentration. The histogram shows the relative intracellular GSH levels (previously normalized to the cell number) in Hep3B cells treated with EFV for 4 h. (C) Mitochondrial membrane potential. TMRM fluorescence in Hep3B cells treated with increasing concentrations of EFV for 1 h, using 10 mM FCCP as a positive control. Data (mean ± SEM, n= 7–11) were calculated as % of control fluorescence value (untreated cells) and analysed by one-way anova multiple comparison test followed by Newman–Keuls test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle). Data for rotenone and FCCP were independently analysed by Student's t-test, § or #P < 0.001.
In keeping with the results of superoxide production, fluorimetric quantification revealed a significant decrease in the intracellular GSH content after 4 h treatment with EFV 25 µM or 50 µM (Figure 4B). Parallel measurements using the fluorescent dye TMRM also revealed a significant decrease in the mitochondrial transmembrane potential (Δψm) after just 1 h treatment with EFV (Figure 4C). A similar decrease in the Δψm was detected at 6 and 24 h (data not shown). The mitochondrial changes induced by 1 h of EFV incubation proved to be reversible, since removal of the drug led to rapid recovery (1 h) of both superoxide production and Δψm to levels similar to those observed in the steady-state (Supporting Information Figure S2).
The mitotoxic effect of EFV was reproduced in human primary hepatocytes, which exhibited both a concentration-dependent increase in mitochondrial superoxide production and a significant decrease in Δψm following incubation with the drug (Figure 5). Importantly, these cells were more sensitive to EFV treatment (1 h) than immortal lines, and the effects observed were detected with lower concentrations of the drug. In particular, Δψm was significantly affected by 10 µM of EFV, a concentration that failed to influence the same parameter in Hep3B cells.
Figure 5.
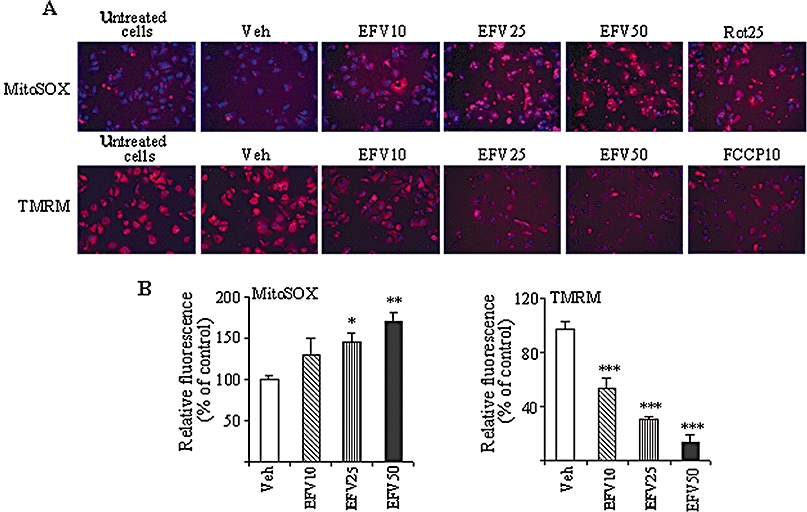
Effect of Efavirenz (EFV) treatment (1 h) on primary human hepatocytes. Rotenone 25 µM or FCCP 10 µM were used as positive controls. (A) Representative images of mitochondrial superoxide production, detected as MitoSOX fluorescence (upper panel: MitoSOX fluorescence in red, Hoechst-stained nuclei shown in blue), and Δψm, detected as TMRM fluorescence (lower panel: TMRM fluorescence in red, Hoechst-stained nuclei shown in blue). (B) Histograms summarizing MitoSOX fluorescence (left panel) and TMRM fluorescence (right panel). Data (mean ± SEM, n= 3–6), calculated as % of control (untreated cells) fluorescence value were analysed by one-way anova multiple comparison test followed by Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle.
EFV treatment induced an increase in mitochondrial mass
Figure 6 shows that incubation (1, 6 and 24 h) with EFV 50 µM, but not 10 or 25 µM, induced a time-dependent increase in the mitochondrial mass measured using the cardiolipin-specific fluorocrome NAO. These results were confirmed with another fluorescent dye, Mitotracker Green FM (results not shown). To further confirm the mitochondrial mass increase induced by EFV, we assessed the mitochondrial protein content. Figure 7A shows the effect of EFV on the expression of several mitochondrial proteins evaluated by Western blot using whole-cell protein extracts where the evaluations were performed after 8 and 24 h of treatment. Although a similar trend was observed in both incubation periods, greater changes were recorded after 24 h of exposure to EFV. The results (Figure 7A) suggest a concentration-dependent increase in the cellular content of Porin, CV subunit α, and β, cyt c and AIF. CIV-subunit II, a mitochondrial DNA-encoded protein, was only slightly and not significantly increased by EFV 50 µM. Interestingly, EFV 50 µM produced a major decrease in the protein expression of all three Complex I subunits studied, CI 17, 20 and 39 kDa, thus pointing to a possible defect in the biogenesis of Complex I. Having detected an increased mitochondrial mass and overall rise in mitochondrial protein content, we next decided to assess the process of mitochondrial biogenesis through analysis of the relative mtDNA copy number. After exposure of Hep3B cells to EFV for 8 and 24 h, the relative copy number of mtDNA normalized to that of nuclear DNA did not present significant changes when compared with vehicle-treated cells (Figure 7B).
Figure 6.
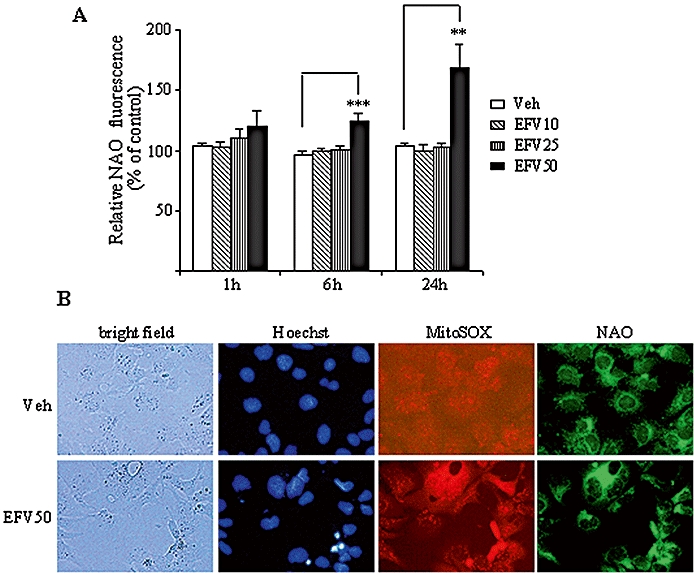
Effect of Efavirenz (EFV) treatment on mitochondrial mass. (A) 10-N-nonyl-acridine orange chloride (NAO) fluorescence in Hep3B cells treated with increasing concentrations of EFV for 1, 6 and 24 h. Data (mean ± SEM, n= 5–9), calculated as % of control (untreated cells) fluorescence were analysed by one-way anova multiple comparison test followed by Newman–Keuls test, **P < 0.01 and ***P < 0.001, versus vehicle. (B) Representative fluorescence microscopy images (20×) of Hep3B cells treated with 50 µM EFV or vehicle (6 h) and stained with Hoechst, MitoSOX and NAO.
Figure 7.
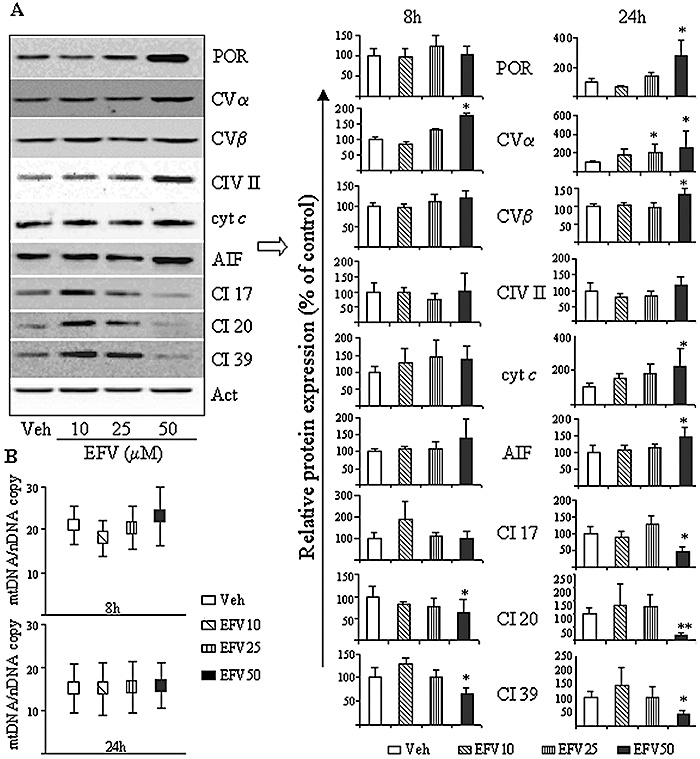
Effect of Efavirenz (EFV) treatment on mitochondrial protein expression. Hep3B cells were treated with increasing EFV concentrations for 8 and 24 h. (A) Representative WB image (24 h) and histograms expressing quantification of the expression of various proteins (8 and 24 h) are shown. Comparisons were made with the protein expression in control (untreated) cells, which was given a relative value of 100% in each individual experiment. Data (mean ± SEM, n= 3–7) were analysed by one-way anova multiple comparison test followed by Newman–Keuls test, *P < 0.05 and **P < 0.01 versus vehicle. (B) Quantitative genomic PCRs used to quantify the relative ratio of mtDNA/nDNA, data represented as mean ± SEM, n= 3–7.
Treatment with Trolox improves the effect of EFV
Figure 8 shows the results obtained with static cytometry experiments performed on Hep3B cells treated with EFV 50 µM for 24 h in the presence of 0.5 mM Trolox. The cytograms in Figure 8A exhibit data from a representative experiment in which Trolox treatment led to a partial recovery of the cell number and partially reversed the increase in chromatin condensation (4′,6-diamino-2-phenylindole channel, left panel) and the increase in mitochondrial mass (FITC channel, right panel) induced by EFV treatment. A summary of the effect of 0.5 mM Trolox on cell survival is provided in Figure 8B.
Figure 8.
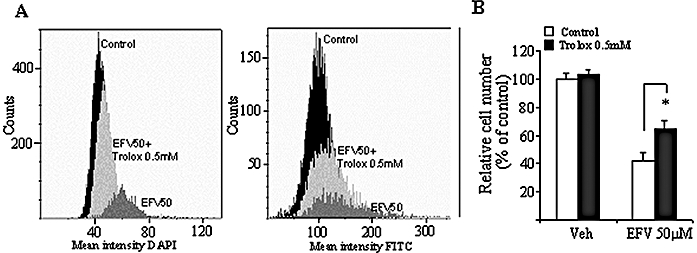
Effect of the antioxidant Trolox on Efavirenz (EFV) 50 µM-treated Hep3B cells. (A) Representative cytograms of untreated control, EFV 50 µM-treated cells and cells treated with EFV 50 µM + 0.5 mM Trolox, showing mean 4′,6-diamino-2-phenylindole (DAPI) (Hoechst) fluorescence (left panel) and mean fluorescein isothiocyanate (FITC) (10-N-nonyl-acridine orange chloride) fluorescence (right panel). (B) Histogram of cell number analysed by static cytometry. Data (mean ± SEM, n= 3–5) were analysed by Student's unpaired t-test, *P < 0.05 versus vehicle.
Discussion
There is growing concern about the side-effects associated with EFV-containing therapies, which include hepatotoxicity. The association between plasma concentration and adverse hepatic effects is well established, and up to 10% of HIV patients treated with EFV exhibit increases in liver enzymes that may require discontinuation of the therapy (Abrescia et al., 2005; Kappelhoff et al., 2005). Furthermore, the potential for hepatotoxicity is known to increase considerably in the presence of HBV/HCV co-infection (Sulkowski et al., 2002; Kontorinis and Dieterich, 2003; Brück et al., 2008), though the reason for this remains elusive. The present study shows that EFV reduces the proliferation and viability of hepatic cells in vitro through an acute mitotoxic effect. These effects were rapid and concentration-dependent, with 10 µM inducing some changes and EFV 50 µM proving to be extremely cytotoxic. Although a static in vitro cell system is clearly different to the in vivo situation, the experimental settings that we employed were chosen to resemble clinical conditions as closely as possible. Thus, we used the human hepatoma cell line Hep3B, which has an active cytochrome P450 system (Zhu et al., 2007), and several experiments were reproduced using primary human hepatocytes. Moreover, the range of concentrations employed was chosen taking into account the important interindividual variability reported for the pharmacokinetics of this drug. Although the recommended daily dose of EFV for adults (600 mg) usually results in plasma levels of 3.17 to 12.67 µM (Starr et al., 1999; Staszewski et al., 1999), several clinical studies have shown that as many as 20% of patients exhibit higher levels, with values of 30–50 µM having been reported (Marzolini et al., 2001; Taylor et al., 2001; Burger et al., 2006). Additionally, there are numerous reports of interactions with a variety of drugs co-administered during the treatment of HIV-1 and which result in a significant increase in the Cmax of EFV (Summary of Sustiva Product Characteristics, 2010). In our study, the inhibitory effects of EFV on proliferation and viability were not specific for hepatic cells and were found to be even more intense in the other two cell lines evaluated (HeLa and HUVEC). These results indicate that the therapeutic effects of EFV are manifested within a narrow range of concentrations and suggest that the drug may be harmful to other tissues.
Our results expand the limited and often contradictory information regarding the effects of EFV on cellular toxicity. For instance, incubation with 12.4 µM EFV for several weeks is reported not to reduce the proliferation of HepG2 cells (Walker et al., 2002). The discrepancy between this finding and our results obtained with Hep3B may be due to metabolic differences between the two hepatoma cell lines (Zhu et al., 2007). On the contrary, 7 days in the presence of 10 or 20 µM EFV results in a significant decrease in the growth of anaplastic thyroid carcinoma ARO cells and poorly differentiated thyroid carcinoma FRO cells (Landriscina et al., 2005). Although these actions have been attributed to changes in the cell cycle, EFV has also been shown to induce caspase- and mitochondria-dependent apoptosis in Jurkat-T and human peripheral blood mononuclear cells in different time frames (Pilon et al., 2002).
There is a direct link between apoptosis and hepatotoxicity (Labbe et al., 2008; Malhi and Gores, 2008; Walters et al., 2009), and there is a growing list of pro-apoptotic compounds associated with liver disorders (Labbe et al., 2008; Malhi and Gores, 2008). In our study, cells treated with 50 µM of EFV displayed clear apoptotic features, such as exposure of phosphatidylserine on the plasma membrane, chromatin condensation, nuclear fragmentation and translocation of the mitochondrial apoptotic proteins cyt c and AIF. In addition, we detected modifications of the cell cycle characteristic of apoptosis in EFV 50 µM-treated cells, namely an increase in the subG1 subpopulation and also G2M arrest. Finally, our evidence points to the involvement of the mitochondrial pathway in the induction of apoptosis, as indicated by the specific activation of caspase-3 and -9 but not of caspase-8 (Danial and Korsmeyer, 2004; Li and Yuan, 2008).
Alterations in mitochondrial physiology and mitochondrial oxidative stress-induced cellular damage are considered to constitute a central event in apoptosis (Valko et al., 2006). Here, we provide evidence of the involvement of mitochondrial dysfunction and oxidative stress in the cellular toxicity produced by EFV. In particular, we observed a significant time- and concentration-dependent increase in mitochondrial superoxide production, which was paralleled by a drop in Δψm and was followed by a reduction in the intracellular GSH content. In addition, treatment with the antioxidant Trolox improved cell survival, a finding that could be of special clinical relevance. These results are in line with those of a recent publication by our group, in which we have reported a direct inhibitory effect of EFV on mitochondrial respiration in both Hep3B and primary human hepatocytes (Blas-García et al., 2010). In that study, measurement of oxygen consumption in isolated mitochondria subjected to EFV revealed that this defect occurred specifically at the level of Complex I of the electron transport chain. Furthermore, the fact that the mitochondrial changes observed in the present study occurred following short exposure to the drug (1 h) suggests that they are unrelated to variations in gene expression or in the number of these organelles, which appear after much longer periods and which, until now, were thought to be the main mechanism of the mitochondrial toxicity of antiretroviral drugs. This hypothesis is reinforced by the fact that the modifications in mitochondrial function that we observed with EFV were reversed soon after removal of the drug.
Some mitochondrial effects were obtained with concentrations of EFV that did not trigger cell death within the time frame of our experiments. It is possible that the effects of the lower EFV concentrations employed in our experiments are accumulative, and would therefore need to be monitored over a longer period of time in order for the damage to become visible. However, it is also feasible that these effects may represent levels of mitochondrial stress below those necessary to induce apoptosis, but which become pro-apoptotic in the presence of other stimuli that further compromise the mitochondria. This is in accordance with the fact that most cases of hepatotoxicity related to EFV coincide with co-morbidities that further interfere with mitochondrial function (Sulkowski et al., 2002). The threshold of EFV-induced mitochondrial stress may also vary depending on the cell type, and it is thus of particular clinical relevance that the effects of EFV were more pronounced in human primary hepatic cells than in Hep3B. Nevertheless, our results do not explain the recent clinical case of an EFV-treated patient who suffered acute liver failure in the apparent absence of mitochondrial damage (Turkova et al., 2009), an adverse effect that could have been related to the co-existence of other risk factors for hepatotoxicity, including idiosyncratic reaction to EFV or the various drugs co-administered with EFV.
Finally, we present data indicating that an increase in mitochondrial mass precedes apoptosis in 50 µM EFV-treated Hep3B cells. This is suggested by the augmented cardiolipin content and enhanced expression of mitochondrial proteins, which was not paralleled by an increase in the mtDNA/nuclear DNA copy number ratio. Interestingly, there are several reports of mitochondrial biogenesis occurring in the absence of an increase in the mtDNA copy number (Lee and Wei, 2005). Moreover, there is mounting evidence of similar increases in mass and/or proliferation of mitochondria in a number of cell types during apoptotic cell death (Mancini et al., 1998; Mahyar-Roemer et al., 2001; Kluza et al., 2004). Although the implication of this increase remains controversial, it is plausible that it constitutes an early response to stress by which the cell adapts to new energy demands and repairs injuries, thus increasing its chances of survival (Lee and Wei, 2005). However, in our case, it was followed by apoptotic cell death, which may be proof that the increased mitochondrial mass in our model was not fully functional. This is in accordance with the progressive accumulation of ROS and the reduction in the protein expression of Complex I subunits. The latter observation, which is in line with our recent finding that EFV directly inhibits Complex I in isolated mitochondria (Blas-García et al., 2010), points to a possible deleterious effect of EFV on Complex I biogenesis. The fact that Trolox partially reversed the effects of EFV on mitochondrial mass further implicates the generation of oxidative stress in this toxic effect of EFV.
In conclusion, the present study demonstrates that EFV is mitotoxic and pro-apoptotic in human hepatic cells in vitro, which may be relevant to the understanding of the hepatotoxicity associated with this drug. Moreover, since EFV is usually administered in conjunction with two NRTI, which are well-known to have deleterious effects on mitochondria, it is tempting to speculate about potential drug combinations in which these and other serious side-effects may be enhanced.
Acknowledgments
The authors thank Dr Jordi Muntane for generously providing the protocol of human hepatocyte extraction and for his assistance with this cell culture, Dr Manuel de Juan for providing liver biopsies, Dr Esteban Martinez for his practical suggestions, Dr Jose Esteban Peris for purifying EFV and for his assistance with the HPLC analysis of EFV solutions, and Brian Normanly for his English language editing of this article.
This work was financed by the Government of Spain grants n° PI081325 (Ministerio de Sanidad y Consumo), SAF2007-60021 (Ministerio de Educación y Ciencia), ACOMP/2009/194 (Conselleria de Educación, Generalitat Valenciana) and CIBER CD06/04/0071, and by an unrestricted grant from Abbott Laboratories.
Glossary
Abbreviations
- AIF
apoptosis-inducing factor
- cyt c
cytochrome c
- DAPI
4′,6-diamino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- EFV
Efavirenz
- FBS
fetal bovine serum
- FCCP
carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone
- FeTCPP
iron(III)-tetrakis (p-carboxyphenyl) porphyrin
- FITC
fluorescein isothiocyanate
- HAART
highly active antiretroviral therapy
- HUVEC
human umbilical vein endothelial cells
- MEM
minimal essential medium
- mtDNA
mitochondrial DNA
- NAO
10-N-nonyl-acridine orange chloride
- NNRTI
non-nucleoside-reverse-transcriptase inhibitor
- NRTI
nucleoside-reverse-transcriptase inhibitor
- Δψm
mitochondrial transmembrane potential
- PI
propidium iodide
- ROS
reactive oxygen species
- STS
staurosporine
- TMRM
tetramethylrhodaminemethylester
- TNF-α
tumour necrosis factor-α
- Trolox
6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid
- WB
Western blotting
Conflict of interest
This work presents no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of EFV on mitochondrial superoxide production. Histogram summarizing data of MitoSOX fluorescence (previously normalized to the cell number) in Hep3B cells treated with EFV for 24 h, detected with a ‘Fluoroscan’ multi-well plate fluorometer. Data (mean ± SEM, n = 5), calculated as % of control (untreated cells) fluorescence value, were analysed by one-way ANOVA multiple comparison test followed by Newman–Keuls test, *P < 0.05, ***P < 0.001 versus vehicle. Data for rotenone were independently analysed by Student's t-test, #P < 0.01.
Figure S2 Effect of EFV removal on mitochondrial function in Hep3B cells. Histograms summarizing data of mean MitoSOX fluorescence (upper panel) and mean TMRM fluorescence (lower panel) in cells treated with increasing concentrations of EFV for 1 h and in cells where EFV was removed after the treatment and fluorescence was recorded 1 h after its removal. Data (mean ± SEM, n = 3–6), calculated as % of control (untreated cells) fluorescence value, were analysed by one-way ANOVA multiple comparison test followed by Newman–Keuls test, *P < 0.05, ***P < 0.001 versus vehicle.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abdul-Ghani MA, DeFronzo RA. Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Curr Diab Rep. 2008;8:173–178. doi: 10.1007/s11892-008-0030-1. [DOI] [PubMed] [Google Scholar]
- Abrescia N, D'Abbraccio M, Figoni M, Busto A, Maddaloni A, de Marco M. Hepatotoxicity of antiretroviral drugs. Curr Pharm Des. 2005;11:3697–3710. doi: 10.2174/138161205774580804. [DOI] [PubMed] [Google Scholar]
- AIDSinfo. A service of the U.S. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. 2010. Available at: http://aidsinfo.nih.gov/Guidelines/Default.aspx?MenuItem=Guidelines (accessed 15 March 2010.
- Blas-García A, Apostolova N, Ballesteros D, Monleón D, Morales JM, Rocha M, et al. Inhibition of mitochondrial function by Efavirenz increases lipid content in hepatic cells. Hepatology. 2010 doi: 10.1002/hep.23647. DOI: 10.1002/hep.23647 ]Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Brück S, Witte S, Brust J, Schuster D, Mosthaf F, Procaccianti M, et al. Hepatotoxicity in patients prescribed efavirenz or nevirapine. Eur J Med Res. 2008;13:343–348. [PubMed] [Google Scholar]
- Burger D, van der Heiden I, la Porte C, van der Ende M, Groeneveld P, Richter C, et al. Interpatient variability in the pharmacokinetics of the HIV non-nucleoside reverse transcriptase inhibitor efavirenz: the effect of gender, race, and CYP2B6 polymorphism. Br J Clin Pharmacol. 2006;61:148–154. doi: 10.1111/j.1365-2125.2005.02536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer S. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Forrest VJ, Kang YH, McClain DE, Robinson DH, Ramakrishnan N. Oxidative stress-induced apoptosis prevented by Trolox. Free Radic Biol Med. 1994;16:675–684. doi: 10.1016/0891-5849(94)90182-1. [DOI] [PubMed] [Google Scholar]
- Gutierrez F, Navarro A, Padilla S, Anton R, Masia M, Borras J, et al. Prediction of neuropsychiatric adverse events associated with long-term efavirenz therapy, using plasma drug level monitoring. Clin Infect Dis. 2005;41:1648–1653. doi: 10.1086/497835. [DOI] [PubMed] [Google Scholar]
- Hammer SM, Eron JJ, Jr, Reiss P, Schooley RT, Thompson MA, Walmsley S, et al. International AIDS Society-USA. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society-USA panel. JAMA. 2008;300:555–570. doi: 10.1001/jama.300.5.555. [DOI] [PubMed] [Google Scholar]
- Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappelhoff BS, van Leth F, Robinson PA, MacGregor TR, Baraldi E, Montella F, et al. 2NN Study Group. Are adverse events of nevirapine and efavirenz related to plasma concentrations? Antivir Ther. 2005;10:489–498. [PubMed] [Google Scholar]
- Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, et al. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene. 2004;23:7018–7030. doi: 10.1038/sj.onc.1207936. [DOI] [PubMed] [Google Scholar]
- Kontorinis N, Dieterich DT. Toxicity of non-nucleoside analogue reverse transcriptase inhibitors. Semin Liver Dis. 2003;23:173–182. doi: 10.1055/s-2003-39948. [DOI] [PubMed] [Google Scholar]
- Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22:335–353. doi: 10.1111/j.1472-8206.2008.00608.x. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landriscina M, Fabiano A, Altamura S, Bagalà C, Piscazzi A, Cassano A, et al. Reverse transcriptase inhibitors down-regulate cell proliferation in vitro and in vivo and restore thyrotropin signaling and iodine uptake in human thyroid anaplastic carcinoma. J Clin Endocrinol Metab. 2005;90:5663–5671. doi: 10.1210/jc.2005-0367. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- Liu Y, Schubert DR. The specificity of neurons by antioxidants. J Biomed Sci. 2009;16:98. doi: 10.1186/1423-0127-16-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahyar-Roemer M, Katsen A, Mestres P, Roemer K. Resveratrol induces colon tumor cell apoptosis independently of p53 and precede by epithelial differentiation, mitochondrial proliferation and membrane potential collapse. Int J Cancer. 2001;94:615–622. doi: 10.1002/ijc.1516. [DOI] [PubMed] [Google Scholar]
- Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M, Sedghinasab M, Knowlton K, Tam A, Hockenbery D, Anderson BO. Flow cytometric measurement of mitochondrial mass and function: a novel method for assessing chemoresistance. Ann Surg Oncol. 1998;5:287–295. doi: 10.1007/BF02303787. [DOI] [PubMed] [Google Scholar]
- Manfredi R, Calza L, Chiodo F. An extremely different dysmetabolic profile between the two available nonnucleoside reverse transcriptase inhibitors: efavirenz and nevirapine. J Acquir Immune Defic Syndr. 2005;38:236–228. doi: 10.1097/01.qai.0000143037.70120.fc. [DOI] [PubMed] [Google Scholar]
- Martin JL, Brown CE, Matthews-Davis N, Reardon JE. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA syntesis. Antimicrob Agents Chemother. 1994;38:2743–2749. doi: 10.1128/aac.38.12.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS. 2001;15:71–75. doi: 10.1097/00002030-200101050-00011. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Pichard L, Raulet E, Fabre G, Ferrini JB, Ourlin JC, Maurel P. Human hepatocyte culture. Method Mol Biol. 2006;320:283–293. doi: 10.1385/1-59259-998-2:283. [DOI] [PubMed] [Google Scholar]
- Pilon AA, Lum JJ, Sanchez-Dardon J, Phenix BN, Douglas R, Badley AD. Induction of apoptosis by a nonnucleoside human immunodeficiency virus type 1 reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2002;46:2687–2691. doi: 10.1128/AAC.46.8.2687-2691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N. Central role of mitochondria in metabolic regulation of liver pathophysiology. J Gastroenterol Hepatol. 2007;22(Suppl 1):S1–S6. doi: 10.1111/j.1440-1746.2007.04963.x. [DOI] [PubMed] [Google Scholar]
- Sebastiá J, Cristofol R, Martin M, Rodriguez-Farre E, Sanfeliu C. Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytometry A. 2003;51:16–25. doi: 10.1002/cyto.a.10003. [DOI] [PubMed] [Google Scholar]
- Starr SE, Fletcher CV, Spector SA, Yong FH, Fenton T, Brundage RC, et al. Combination therapy with efavirenz, nelfinavir, and nucleoside reverse-transcriptase inhibitors in children infected with human immunodeficiency virus type 1. Pediatric AIDS Clinical Trials Group 382 Team. N Engl J Med. 1999;341:1874–1881. doi: 10.1056/NEJM199912163412502. [DOI] [PubMed] [Google Scholar]
- Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, et al. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. Study 006 Team. N Engl J Med. 1999;341:1865–1873. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- Sulkowski MS, Thomas DL, Mehta SH, Chaisson RE, Moore RD. Hepatotoxicity associated with nevirapine or efavirenz-containing antiretroviral therapy: role of hepatitis C and B infections. Hepatology. 2002;35:182–189. doi: 10.1053/jhep.2002.30319. [DOI] [PubMed] [Google Scholar]
- Summary of Sustiva Product Characteristics. Bristol-Myers Squibb. 2010. http://packageinserts.bms.com/pi/pi_sustiva.pdf (assessed on 15 March 2010.
- Tashima KT, Bausserman L, Alt EN, Aznar E, Flanigan TP. Lipid changes in patients initiating efavirenz- and indinavir-based antiretroviral regimens. HIV Clin Trials. 2003;4:29–36. doi: 10.1310/f2v7-3r46-vx6j-241r. [DOI] [PubMed] [Google Scholar]
- Taylor S, Reynolds H, Sabin CA, Drake SM, White DJ, Back DJ, et al. Penetration of efavirenz into the male genital tract: drug concentrations and antiviral activity in semen and blood of HIV-1-infected men. AIDS. 2001;15:2051–2053. doi: 10.1097/00002030-200110190-00022. [DOI] [PubMed] [Google Scholar]
- Turkova A, Ball C, Gilmour-White S, Rela M, Mieli-Vergani G. A paediatric case of acute liver failure associated with efavirenz-based highly active antiretroviral therapy and effective use of raltegravir in combination antiretroviral treatment after liver transplantation. J Antimicrob Chemother. 2009;63:623–625. doi: 10.1093/jac/dkn548. [DOI] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Walker UA, Setzer B, Venhoff N. Increased long-term mitochondrial toxicity in combinations of nucleoside analogue reverse-transcriptase inhibitors. AIDS. 2002;16:2165–2173. doi: 10.1097/00002030-200211080-00009. [DOI] [PubMed] [Google Scholar]
- Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM, et al. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLOS Pathog. 2009;5:e1000269. doi: 10.1371/journal.ppat.1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Ping YH, Yen JC, Chang CY, Wang SF, Yeh CL, et al. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol. 2007;220:243–251. doi: 10.1016/j.taap.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Zhu XH, Wang CH, Tong YW. Growing tissue-like constructs with Hep3B/HepG2 liver cells on PHBV microspheres of different sizes. J Biomed Mater Res B Appl Biomater. 2007;82:7–16. doi: 10.1002/jbm.b.30698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.