

Abstract
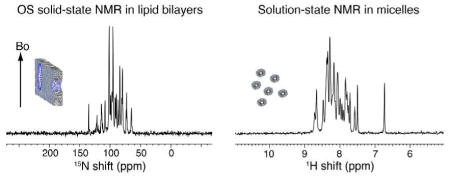
A mixture of phospholipids and Triton X-100 at a molar ratio of 5 (q=5) forms well aligned and stable bilayers that give superior solid-state NMR spectra of proteins. In this comparison, the solid-state NMR spectrum of Pf1 coat protein in aligned phospholipid bilayers (left) displays better resolution than the equivalent solution NMR spectrum (right) of the same protein in micelles. Both samples and experimental parameters were fully optimized.
Molecular alignment is an integral part of solution NMR of soluble proteins and solid-state NMR of membrane proteins in phospholipid bilayers. Starting with Pake’s demonstration that the splittings between doublets due to the dipole-dipole interaction depend on the angle with respect to the field,1 there is a long history of devising ways to measure these splittings, the most powerful of which is separated local field (SLF) spectroscopy, in part because it is equally applicable to single crystals2 and uniaxially aligned3 samples. Once it was shown that the natural tendency of proteins in solution to align in a strong magnetic field could be boosted by the use of alignment media, such as bicelles4, filamentous bacteriophages5, or stressed polyacrylamide gels6 it became possible to routinely measure residual dipolar couplings (RDCs) for structure determination and refinement, including of small membrane proteins in micelles or isotropic bicelles. Similarly, once it was shown that membrane proteins in phospholipid bilayers could be aligned magnetically in mixtures of long chain and short chain lipids to an extent that was superior to that obtainable by mechanical alignment between glass plates7, it became possible to routinely obtain solid-state NMR spectra with single-site resolution and directly measure individual dipolar couplings as input for structure calculations.8 Because of the importance and widespread implementation of dipolar coupling measurements, there has been continuous development of the alignment media for use with both soluble and membrane proteins.
Here we report an advance in sample preparation; by increasing the resolution of the spectra and the stability of the samples it markedly improves the applicability of oriented sample (OS) solid-state NMR to structure determination of membrane proteins in phospholipid bilayers. Mixtures of “long chain lipids”, phosphatidyl choline lipids whose hydrophobic chains vary in length and unsaturation, with the “short chain lipid” Triton X-100 in a molar ratio of 5:1 (q=5) form planar magnetically alignable bilayers9 that are stable over a wide temperature range for long periods of time (months) and yield protein spectra whose resolution and sensitivity are superior to those we have obtained in any other combination of lipids.
This is illustrated in Figure 1 with one-dimensional 15N solid-state NMR spectra of uniformly 15N labeled membrane proteins with one, two, and seven trans-membrane helices, and corresponding 31P NMR spectra of the phospholipids in the same samples. As is the case for conventional DMPC:DHPC bicelles (q>2.5), the bilayer normal is perpendicular to the direction of the applied magnetic field, and narrow single-line resonances are observed because both the lipids and the proteins undergo fast rotational diffusion about the bilayer normal. It is also possible to “flip” the direction of alignment to parallel to the field through the addition of lanthanide ions (Figure S1).10
Figure 1.
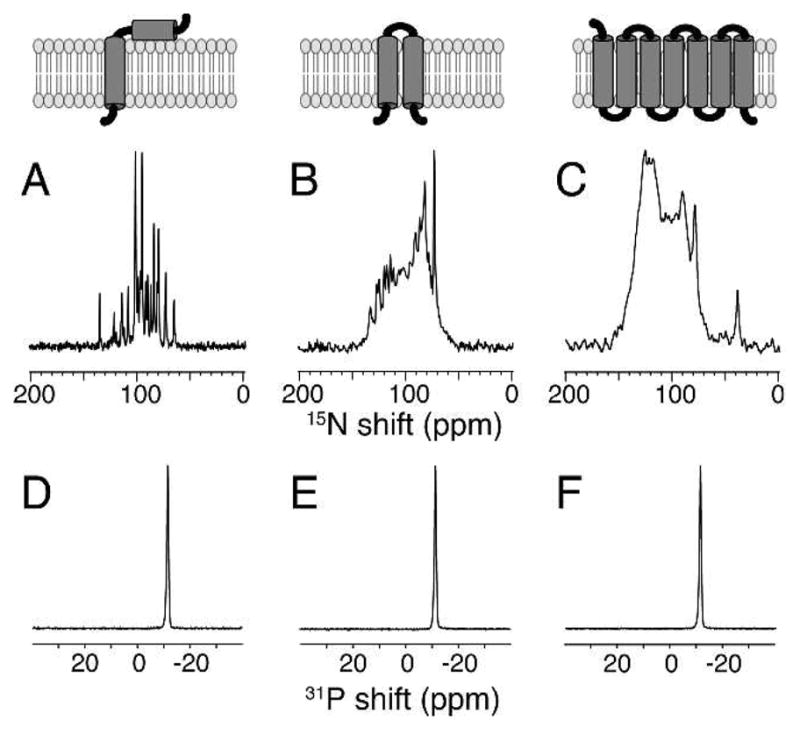
The top row contains schematic drawings of the three membrane proteins that are aligned in planar “long chain lipids”:Triton X-100 (q=5) bilayers. (A–C) One-dimensional solid-state 15N NMR spectra of uniformly 15N labeled proteins. (D–F) One-dimensional solid-state 31P NMR spectra of the phospholipids. (A and D) The 46-residue Pf1 coat protein. (B and E) The 78-residue mercury transport protein MerE. (C and F) The 350-residue G-protein coupled receptor CXCR1. The 15N NMR spectra were obtained at 35°C on a Bruker 700 MHz spectrometer using a homebuilt 1H/15N double-resonance probe with a MAGC coil for the 1H channel and a solenoid coil for the 15N channel12. Pf1 coat protein and MerE are aligned in DMPC:Triton X-100 bilayers, and CXCR1 is aligned in DMPC:POPC (8:2, w/w):Triton X-100 bilayers.
All of the spectra in Figure 1 are distinguished by the narrow line widths of their resonances. There is only a single narrow (<1 ppm) 31P resonance from the phospholipids; the resonance frequency demonstrates the perpendicular alignment and planarity of the DMPC bilayers. The narrow (~0.5 ppm) line widths of the 15N resonances from the amide backbone sites in the proteins are most readily appreciated in Figure 1A where essentially every signal is resolved. Although there is significant overlap in the spectra in Figure 1B and 1C from the larger proteins, the sharp features of the overlapped resonances indicate that the line widths of individual resonances are similar to those in Figure 1A.
For simplicity we show half of the symmetric SLF spectra in Figure 2, thus each dipolar coupling is actually twice the value observed for each of the backbone amide 1H-15N dipolar couplings. Except for the coincident resonances of residues 24 and 28, this two-dimensional spectrum is fully resolved. It is qualitatively superior to earlier versions11 (Figure S2) and this improvement can be ascribed solely to the superior alignment and stability of the DMPC:Triton X-100 bilayers. The protein preparation, spectrometer, probe, and pulse sequence are essentially identical to those used previously.
Figure 2.
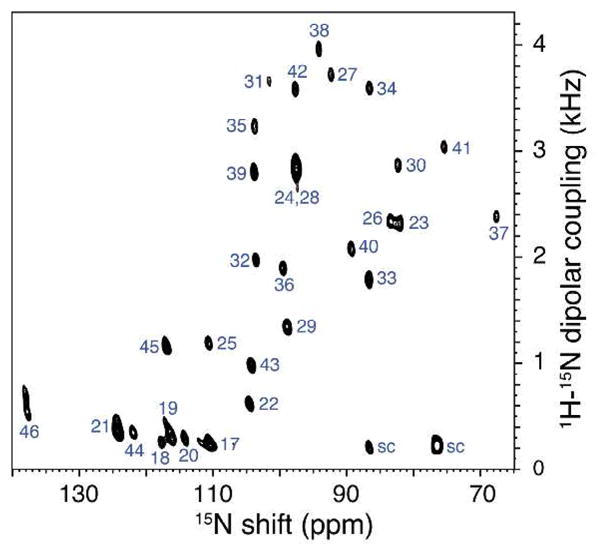
Two-dimensional SLF spectrum of uniformly 15N labeled Pf1 coat protein in DMPC:Triton X-100 bilayers obtained at 700 MHz with the SAMPI4 pulse sequence13. It resulted from signal averaging 64 transients for each of 128 t1 points, and 1024 points were acquired in the direct dimension. The final data matrix had 1024 by 1024 points.
Phospholipid bilayers align in the magnetic field when a short change phospholipid (e.g. DHPC) or a detergent (e.g. CHAPSO or Triton X-100) is present. These alignment reagents dissolve in the long-chain phospholipid bilayers, and typically reduce the order parameter of both the phospholipids and the proteins to 0.8–0.9 compared to 1.0 observed in DMPC bilayers. Above a certain concentration they also cause defects in the bilayer. Since it is possible under some circumstance to form aligned bilayers with a q as high as 10 where no separate 31P signal can be observe for the short chain lipids that ‘cap’ the defects or rims of the bilayer discs, it may be that the dissolution of DHPC or the detergent into the long-chain bilayers is sufficient to induce magnetic alignment. The 31P NMR signal from the long-chain phospholipids is routinely used as measure of the alignment, since its frequency and width vary significantly with added protein and variation of experimental parameters especially temperature. As shown in Figure 1, for membrane proteins with between 46 and 350 residues, the 31P NMR signal from the phospholipids is extremely narrow, indicative of very well aligned planar bilayers. In favorable cases it is possible to obtain similar line widths from DMPC:DHPC samples, however, the quality of the 15N NMR spectra of the proteins in these samples do not approach those shown in Figure 1. Thus, we believe that there is a second factor that contributes to the success of DMPC:Triton X-100 bilayers, and that is the absence of a strong temperature dependence on alignment. Even when minimized through the use of “low E” coils, it is still difficult to avoid a few degrees of sample heating in the course of signal averaging under conditions of high power radio-frequency irradiation at high frequencies. With other bilayer preparations, this relatively small temperature variation is sufficient to shift and broaden the resonances of both the 31P and 15N NMR signals. Indeed, part of the set up of experiments using DMPC:DHPC mixtures is to empirically vary the sample temperature by a few degrees to obtain the narrowest line widths. As shown in Figure S3 of the Supplemental Material, DMPC:Triton X-100 bilayers remain well aligned over a broad range of temperatures. This explains why neither the 31P nor the 15N NMR signals are broadened by the few degrees of sample heating inherent in these experiments. This effect may be just as important as the fundamentally high degree and uniformity of the magnetic alignment of the samples.
The combination of narrow line widths in both the dipolar coupling and chemical shift dimensions in two-dimensional spectra, and the long-term stability of the samples improves the prospects for resolving signals from individual sites in membrane proteins with multiple trans-membrane helices. Although the one-dimensional spectra in Figure 1B and 1C display limited resolution, this is due to the overlap of many narrow lines, which can be resolved in multidimensional experiments, rather than fundamentally broad line widths. This furthers the application of OS solid-state NMR to membrane proteins, since there is no fundamental reason for the line widths in a large membrane protein to be broader than those in small membrane protein in phospholipid bilayers
Supplementary Material
Acknowledgments
We thank Woo Sung Son for the sample of MerE and his advice, and Fabio Casagrande, Mignon Chu, Klaus Maier, and Hans Kiefer for their assistance with the preparation of the CXCR1 sample. This research was supported by grants from the National Institutes of Health, and utilized the Biomedical Technology Resource for NMR Molecular Imagining of Proteins at UCSD.
Footnotes
Supporting Information Available: Experimental details, and 15N and 31P NMR spectra of a DMPC:Triton X-100 sample at various temperatures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pake GE. J Chem Phys. 1948;16:327–331. [Google Scholar]
- 2.Hester RK, Ackerman JL, Neff BL, Waugh JS. Phys Rev Lett. 1976;36:1081–3. [Google Scholar]
- 3.Opella SJ, Waugh JS. J Chem Phys. 1977;66:4919–24. [Google Scholar]
- 4.Tjandra N, Bax A. Science. 1997;278:1111–4. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 5.(a) Clore GM, Starich MR, Gronenborn AM. J Am Chem Soc. 120:10571–2. [Google Scholar]; (b) Hansen MR, Hanson P, Pardi A. Methods Enzymol. 2000;317:220–40. doi: 10.1016/s0076-6879(00)17017-x. [DOI] [PubMed] [Google Scholar]; (c) Park SH, Son WS, Mukhopadhyay R, Valafar H, Opella SJ. J Am Chem Soc. 2009;131:14140–1. doi: 10.1021/ja905640d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. J Biomol NMR. 2000;18:303–9. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]; (b) Tycko R, Blanco FJ, Ishii Y. J Am Chem Soc. 2000;122:9340–9341. [Google Scholar]; (c) Jones DH, Opella SJ. 2004;171:258–69. doi: 10.1016/j.jmr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 7.De Angelis AA, Nevzorov AA, Park SH, Howell SC, Mrse AA, Opella SJ. J Am Chem Soc. 2004;126:15340–1. doi: 10.1021/ja045631y. [DOI] [PubMed] [Google Scholar]
- 8.De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. J Am Chem Soc. 2006;128:12258–67. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sanders CR., 2nd Biophys J. 1993;64:171–81. doi: 10.1016/S0006-3495(93)81352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sanders CR, 2nd, Schaff JE, Prestegard JH. Biophys J. 1993;64:1069–80. doi: 10.1016/S0006-3495(93)81473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prosser RS, Hunt SA, DiNatale JA, Vold RR. J Am Chem Soc. 1996;118:269–70. [Google Scholar]
- 11.Opella SJ, Zeri AC, Park SH. Annu Rev Phys Chem. 2008;59:635–57. doi: 10.1146/annurev.physchem.58.032806.104640. [DOI] [PubMed] [Google Scholar]
- 12.Grant CV, Yang Y, Glibowicka M, Wu CH, Park SH, Deber CM, Opella SJ. J Magn Reson. 2009;201:87–92. doi: 10.1016/j.jmr.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nevzorov AA, Opella SJ. J Magn Reson. 2007;185:59–70. doi: 10.1016/j.jmr.2006.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.