

Abstract
The purpose of this study was to test the hypothesis that mineral maturity and crystallinity index are two different characteristics of bone mineral. To this end, Fourier Transform InfraRed Microspectroscopy (FTIRM) was used. To test our hypothesis, synthetic apatites and human bone samples were used for the validation of the two parameters using FTIRM. Iliac crest samples from seven human controls and two with skeletal fluorosis were analyzed at the bone structural unit (BSU) level by FTIRM on 2–4 μm-thick sections. Mineral maturity and crystallinity index were highly correlated in synthetic apatites, but poorly correlated in normal human bone. In skeletal fluorosis, crystallinity index was increased and maturity decreased, supporting the fact of separate measurement of these two parameters. Moreover, results obtained in fluorosis suggested that mineral characteristics can be modified independently of bone remodeling. In conclusion, mineral maturity and crystallinity index are two different parameters measured separately by FTIRM and offering new perspectives to assess bone mineral traits in osteoporosis.
Keywords: Apatites; chemistry; Bone and Bones; chemistry; Calcification, Physiologic; physiology; Humans; Spectroscopy, Fourier Transform Infrared; X-Ray Diffraction
Keywords: Bone mineral, Hydrated layer, FTIRM, Mineral maturity, Mineral crystallinity index
Introduction
Human bone mineral is constituted of a poorly crystallized apatite. It is a calcium (Ca)-deficient apatite, containing hydrogen phosphate (HPO4), carbonate (CO3) and other ions. The hydroxyapatite crystal structure belongs to the hexagonal system. The surface of bone crystal, formed in the water of extracellular fluid (ECF), exhibits a “hydrated layer” (Fig. 1). Ions in this layer are very labile and reactive, and constitute the non-apatitic domain, surrounding the relatively inert and more stable apatite domain of the bone crystal [1, 2]. Newly deposited bone mineral contains many labile non-apatitic domains [HPO4, phosphate (PO4), and CO3], which are located in the well-developed hydrated layer involved in the high surface reactivity of mineral [3]. Labile PO4 and CO3 groups are easily and reversibly exchangeable with each other in the hydrated layer. During maturation, the decrease in labile non-apatitic environments is associated with an increase in stable apatitic environments [3]. A particularity of the bone mineral is its non-stoichiometry, leading to the presence of numerous vacancies in the apatite crystal. Consequently, bone crystal cohesion is mainly maintained by electrostatic cohesion, thus bone crystals are easily soluble relative to stoichiometric apatite [4]. As bone becomes more mature, both the size and number of crystals increase.
Figure 1.
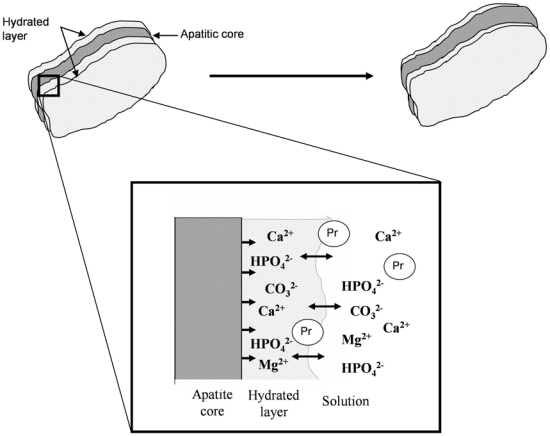
Evolution of the hydrated layer and crystal apatite. During the maturation and growth of the crystal, the hydrated layer, involved in a high surface reactivity, progressively decreased and led to a stable apatitic domain. The structure of the hydrated layer constitutes a pool of loosely bound ions which can be incorporated in the growing apatite domains and can be exchanged by foreign ions in the solution and charged groups of proteins (Pr). Courtesy of C. Rey (Rey et al. (2009); Osteoporos. Int :1013–1021)
Vibrational spectroscopy techniques, such as Fourier Transform InfraRed Spectroscopy (FTIRS), Synchrotron InfraRed or Raman Spectroscopy, have been extensively used to study calcified tissues [3, 5–13]. Spectroscopic techniques allow assessment of physicochemical modifications of mineral induced by mechanical tests [5, 14–17], age-related modifications [5, 6, 14, 18], and pathologic or treatment-related changes [7, 19–23]. The application of Fourier Transform InfraRed Microspectroscopy and Imaging (FTIRM, FTIRI) for bone allows in situ analysis of embedded bone samples at the bone structural unit (BSU) level.
The purpose of this study was to test the hypothesis that mineral maturity (transformation of non apatitic domains into apatitic ones) and mineral crystallinity index (size/strain and perfection) are two different characteristics of bone mineral. These two parameters are temporally linked and often well correlated in synthetic apatite. However, they can evolve separately in human bone, and thus can independently affect the mineral characteristics. Indeed, mineral maturity can be affected by modification of bone remodeling (and by formative or antiresorptive treatments), whereas crystallinity can be influenced by ionic substitutions. To test this hypothesis, bone samples from two patients with skeletal fluorosis were analyzed. Skeletal fluorosis is a pathology caused by an excessive consumption of fluoride, and characterized by ionic substitution of hydroxyl ions by fluoride ions in bone mineral. Each parameter was first validated using synthetic apatites and/or control bone, and then measured on seven human samples from adult controls. Thus, the main purpose of the present study was to determine, at the BSU level, if we can distinguish two distinct parameters involved in bone mineral characteristics, i.e., mineral maturity and mineral crystallinity index.
Materials and methods
Synthetic apatites
Six non-carbonated nanocrystalline apatites corresponding to different maturation periods (0, 1, 3, 15 and 60 days), and with known HPO4 content (22%, 22.8%, 18.4%, 13%, 9.8% respectively), were prepared as follows: nanocrystalline biomimetic apatites were prepared by double decomposition in aqueous media, at physiologic pH, between a solution of calcium nitrate (Ca(NO3)2, 4H2O : 17.7 g for 250 ml of de-ionized water) and an ammonium hydrogen phosphate solution (NH4)2HPO4 : 40 g for 500 ml of de-ionized water). The calcium solution was poured rapidly into the phosphate solution. The excess of phosphate ions has a buffering effect and allows the pH to stabilize around 7.4 without the use of any foreign molecules or ions [24]. The suspensions were left to age at room temperature for variable periods of time (maturation). They were then filtered on a Büchner funnel and washed with de-ionized water until elimination of ammonium and nitrate counter ions. The samples were then freeze-dried and stored at low temperature (−18°C) to prevent any further alteration. The main constituents of the precipitated apatites were analyzed. The calcium content was determined by complexometry with EDTA, the total phosphate content by UV-vis spectrophotometry of the phosphovanadomolybdic complex and the HPO42− fraction was determined using the method proposed by Gee and Dietz, involving the thermal condensation of hydrogen phosphate ions into pyrophosphate [25] and the dosage of pyrophosphate groups. All samples were analyzed by X-ray diffraction using a curved counter (INEL CP 120, Co Kα radiation). They presented a pattern characteristic of poorly crystalline apatites, analogous to those observed for biologic samples (not shown).
Three carbonated apatites (maturation time: 1, 6 and 30 days) containing 3.95%, 5.10% and 5.69%, respectively, of CO3 were also used [analyses of CO3 were made by coulometry (Coulometrics CM 5130, UIC inc.)].
Between 0.7–1% of synthetic apatite was mixed with KBr, compressed into pellets, and then analyzed by FTIR macrospectroscopy.
Human bone samples
Seven iliac crest bone samples from adult controls (43–93 years old) without apparent metabolic bone disease were obtained at necropsy (Université Claude Bernard, Lyon, France). Iliac crest bone samples from two patients with skeletal fluorosis were also analyzed. Skeletal fluorosis was diagnosed on clinical, radiologic, and histomorphometric criteria, and confirmed using a chemical dosage of fluoride with a specific ion electrode. Bone samples were fixed in alcohol, dehydrated in absolute alcohol, and then embedded in methylmethacrylate [26]. Non-decalcified sections (2–5 μm thick) were cut with a microtome Polycut E (Reichert-Jung (Leica), Germany).
FTIR Spectroscopy and Microspectroscopy
For FTIR spectroscopy performed on KBr pellets, a dried-air purge system was used to minimize air and CO2 bands. For each pellet, 20 scans were collected at 4 cm−1 resolution in the transmission mode and analyzed with a Fourier Transform Infrared Microspectroscope (Perkin-Elmer GXII Auto-image Microscope) equipped with a wideband detector (mercury-cadmium-telluride) (7800–400 cm−1). After baseline correction by automatic correction and normalization at absorbance 1.5 on the υ3PO4 (Spectrum Software), the curve fitting of every individual spectrum was performed.
FTIRM was performed on 2–5 μm-thick sections. Each spectrum was collected at 4 cm−1 resolution and 200 scans in the transmission mode. The instrument used an objective Cassegrain of numerical aperture 0.6; the system has a spatial resolution of 10 μm at typical mid-infrared wavelengths. Contribution of air and MMA were subtracted from the original spectrum. Each spectrum was baseline corrected and normalized at absorbance 1.5 on the υ3PO4. For the seven control human bone samples, spectra of 5–10 areas of 50×50 μm were acquired separately in newly formed and old interstitial bone, in both cortical and cancellous bone, with a total of 20–40 measurements for each sample. Selection of newly formed and old bone was based on morphologic criteria. The new bone is the surface bone, and the old one is the interstitial bone. Because a great part of the old bone is quiescent in surface, the new bone is differentiated according to its low mineralization index. In patients with a double tetracycline labeling, the sections were observed under fluorescent light to identify newly formed bone.
GRAMS/AI software (Thermo Galactic, Salem, NH, USA) was used to quantify the characteristics of the spectra. For the phosphate vibration, we used synthetic hydroxyapatites, carbonated apatites and previously published data [13]. Concerning ν1ν3PO4 vibration, five sub-bands were used (1110, 1082, 1060, 1030, 962 cm−1), and for the ν4PO4 vibration, four sub-bands were used (604, 577, 563, 552 cm−1). After curve-fitting of every individual spectrum, position, height, full width at half-maximum (FWHM), and area under the curves were measured. Peaks corresponding to ν1ν3PO4 (900–1200 cm−1) and ν4PO4 (500–650 cm−1) were analyzed. The following parameters were calculated: (1) mineral maturity (1030/1110 cm−1 area ratio); (2) mineral crystallinity index (defined as being inversely proportional to the FWHM of 604 cm−1 peak). The peak located at 604 cm−1 is of particular interest because of its good resolution. It corresponds to apatitic phosphate environment, and gives direct information on the crystallinity index of the sample. The narrower the peak is, the higher the crystallinity index. In order to validate the using of the width at half-height of 604 cm−1 as a crystallinity index by infrared spectroscopy, this latter has been compared on same samples with the crystallinity index measured by the Shemesh method [27], derivated from the splitting factor measured by Termine and Posner [28]. This crystallinity index [29] corresponds to the splitting of a triply degenerate antisymmetric bending vibration of orthophosphate and to the following ratio: CI=(A604+A564)/A590, where Ax is the absorbance at wavenumber x (Fig. 2A).
Figure 2.
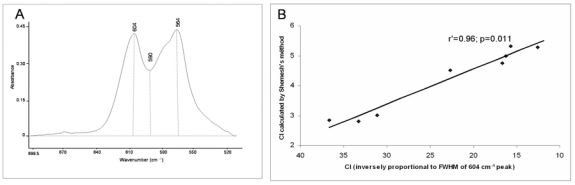
Synthetic apatites, ν4PO4vibration (A) Crystallinity index [29] measured by Shemesh’s method [29], given by CI = {A604 + A564}/A590. (B) Correlation between the CI measured by the Shemesh’s method and by FTIRM in which CI is inversely proportional to the full width at half maximum (FWHM) of the 604 cm−1 peak.
X-ray diffraction (XRD)
XRD, used to validate the measurements of crystallinity performed by FTIRM, was done at the Centre de Diffractométrie Henri Longchambon (Université de Lyon, France). Powdered synthetic samples were analyzed using a Bruker® D8 Advance diffractometer equipped with a PSD Detector (VÅNTEC-1 “Super Speed”). Profiles were obtained between 0 and 70° 2θ, and a count of 2 s was performed at room temperature every 0.2°. Two different crystallographic directions were analyzed: the (002) and (310) reflections. The (002) reflection was related to the length axis (c-axis), and (310) reflection was related to the dimension perpendicularly to the c-axis (crystal width). For bone samples, thick sections of bone biopsies were analyzed by XRD. Two pairs of measurement were done, with one fluorotic bone and one control bone, age- and sex-matched for each pair. XRD analyses were performed between 20 and 45° 2θ, with a timescale of 40 (equivalent to a measurement on a scintillation counter with a step of 2/100 and an exposure time of 40 s/step). The FWHM of (002) and (310) reflections were analyzed after curve-fitting with TOPAS P software (Bruker®) The narrower the peak is, the higher the crystallinity.
Statistical analysis
Non-parametric tests were used because of the small number of samples. Four regions of interest were studied: new and old cortical bone, and new and old cancellous bone. Data were analyzed using two-way analysis of variance (Friedman Anova). If differences were significant, Wilcoxon’s T-test was used to compare either cortical versus cancellous, or new versus old bone. Spearman correlations between methods and between parameters were performed. A test is considered significant if p≤0.05.
Results
Validation of the characteristics of mineral
A part of a spectrum obtained from a synthetic apatite in the region corresponding to PO4 and CO3 vibrations is illustrated in Figure 3.
Figure 3.
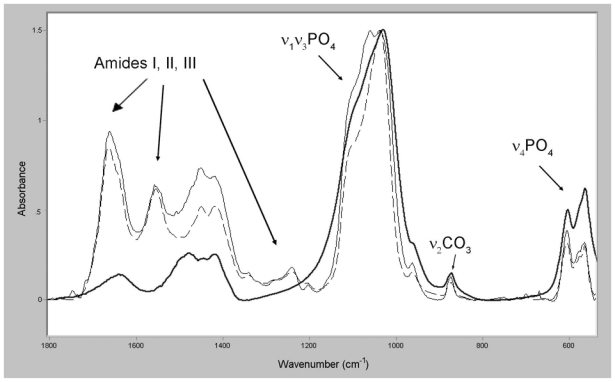
Infrared spectra obtained from (1) a synthetic carbonated apatite and showing the ν1ν3PO4, ν2CO3 and the ν4PO4 vibrations (bold curve), (2) new (full curve) and old (dotted curve) bone showing different vibrations, including those of amides (I, II and III).
Mineral maturity
Mineral maturity represents the progressive transformation of non-apatitic domains into poorly then well-crystallized apatite (Fig. 1). This parameter is measured as the evolution of the ν1ν3PO4 vibration in synthetic apatites of known state of maturation (Fig. 4A). The variation of the content of HPO4 ions essentially located in the hydrated layer reflects its evolution. The 1030 cm−1 peak has been assigned to apatitic phosphate groups and is observed in well-crystallized stoichiometric hydroxyapatite [13]. The 1110 cm−1 peak is present in nanocrystalline apatites and it has been assigned to non-apatitic phosphate or HPO4 in poorly crystalline apatite [13, 30, 31]. Thus, the area ratio 1030/1110 cm−1 gives an index of mineral maturity corresponding to the transformation of a non-apatitic domain into apatitic ones. In synthetic apatites, this area ratio increases progressively with mineral maturation (Fig. 4B) and is inversely correlated with the HPO4 content (r2=0.89, p<0.02).
Figure 4.
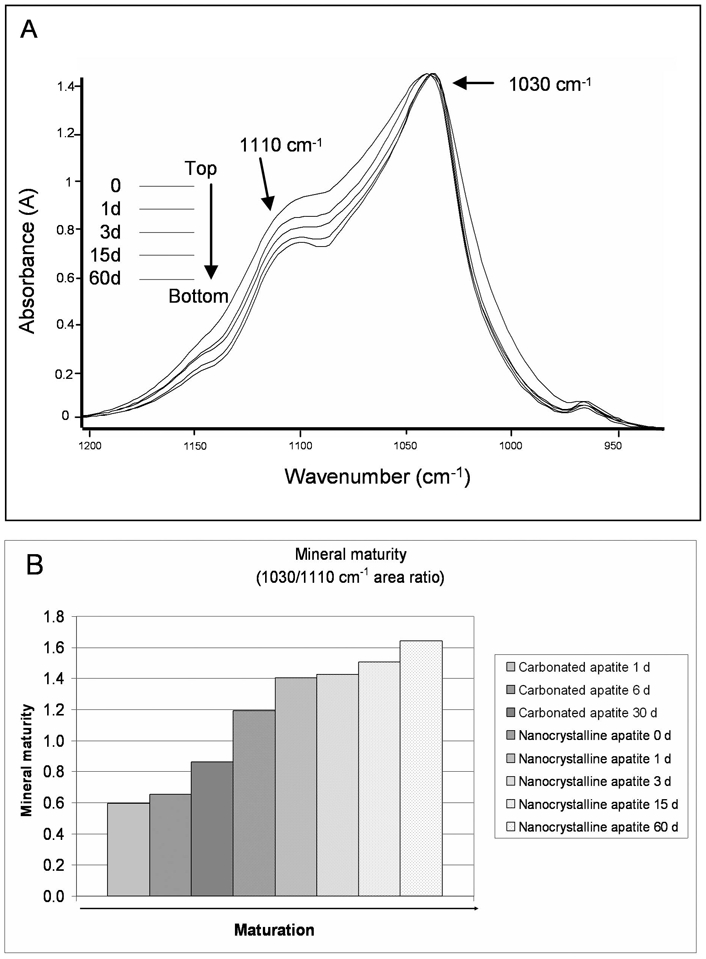
Evolution of mineral maturity (ν1ν3PO4) vibration in various synthetic apatites. (A) Non-apatitic phosphate (1110 cm−1) decreases with the progression of mineral maturation, while the apatitic phosphates (1030 cm−1) are constant. (B) Mineral maturity increases with the progression of maturation (d: days of maturation).
Mineral crystallinity index
Mineral crystallinity is defined as the degree of order in a solid as measured by XRD. Validation of the crystallinity index values was done first by comparing measurements done by FTIRM (inversely proportional to the FWHM of the 604 cm−1 peak assigned to phosphate ions in the apatite domains) and the ratio used by Shemesh et al [27]. Results have shown that there is an excellent correlation between the two crystallinity indexes measured by FTIR (Fig. 2B; r′=0.96, p=0.011). Then, our crystallinity index (inversely proportional to the FWHM of the 604 cm−1 peak) has been compared with measurements done by XRD (FWHM of (002) and (310) apatite reflections). A good and significant correlation was found (r′=0.93, p=0.0083; r′=0.91, p=0.016, respectively) in synthetic apatites between the crystallinity parameters measured by FTIRM and by XRD (Figs. 5A and 5B).
Figure 5.
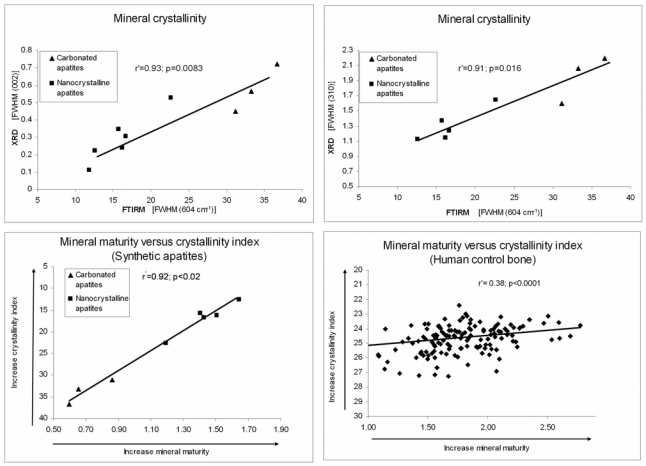
Evolution of mineral crystallinity and correlation between crystallinity index and mineral maturity in synthetic apatites. (A) Significant correlation between the crystallinity index measured by FTIRM (604 cm−1 peak) and the crystallinity measured by X-ray diffraction [XRD, (002) reflection]. (B) Significant correlation between the crystallinity index measured by FTIRM (inversely proportional to the FWHM of 604 cm−1 peak) and the crystallinity measured by the XRD (310) reflection. (C) Correlation between mineral maturity and mineral crystallinity index in synthetic apatites. These two parameters are closely linked (FWHM: full width at half maximum). (D) Correlation between mineral maturity and mineral crystallinity index in human bone.
Correlation between mineral maturity and crystallinity index in synthetic apatites
In synthetic apatites (Fig. 5C), mineral maturity and crystallinity index were highly correlated (r′=0.92, p<0.02) and thus temporally linked.
Bone mineral from human controls
The spectra obtained from new and old bone in human adult controls (Fig. 3) illustrated differences in mineral associated with the aging of bone. For all four bone areas, differences were significant (p=0.0014–0.0056). Comparisons between cortical versus cancellous, or new versus old bone are shown in Table 1.
Table 1.
Mineral maturity and mineral crystallinity index measured in iliac bone samples from human adult controls and from patients with skeletal fluorosis.
| Mineral maturity (Area ratio 1030/1110 cm−1) Mean (SD) | Mineral crystallinity index (inversely proportional to FWHM 604 cm−1) Mean (SD) | |
|---|---|---|
| Control bone (n: 7) | ||
| Total new bone | 1.643 (0.265) | 25.078 (0.923) |
| Total old bone | 1.926a (0.274) | 24.536b (0.641) |
| Cortical new bone # | 1.738 (0.280) | 25.166 (0.693) |
| Cancellous new bone # | 1.549 (0.230) | 24.990 (0.994) |
| Cortical old bone # | 2.046 (0.271) | 24.636 (0.693) |
| Cancellous old bone # | 1.806c (0.236) | 24.436 (0.994) |
| Fluorotic bone (n: 2)§ | ||
| Cortical new bone | 1.293 (0.056) | 20.377 (1.711) |
| Cancellous new bone | 1.185 (0.078) | 20.170 (0.670) |
| Cortical old bone | 1.534 (0.187) | 19.062 (1.302) |
| Cancellous old bone | 1.422 (0.047) | 19.131 (0.868) |
Human bone
Mean of 5–10 measurements per sample in each compartment.
Friedman Anova, p: 0.0014–0.0056
Wilcoxon’s T-test: Comparison of new bone versus old bone,
: p=0.001;
: p <0.006
Wilcoxon’s T-test: Comparison of cortical new versus cancellous new bone: NS
Wilcoxon’s T-test: Comparison of cortical old versus cancellous old bone,
: p<0.03
Fluorotic bone
For each sample, mean of 5–10 measurements per sample in each compartment.
FWHM: Full Width at Half Maximum; NS: not significant; SD: standard deviation
Mineral maturity
The 1110 cm−1 peak was less intense in old interstitial bone than in newly formed bone (young BSUs) (Figs. 6A and 6B).
Figure 6.
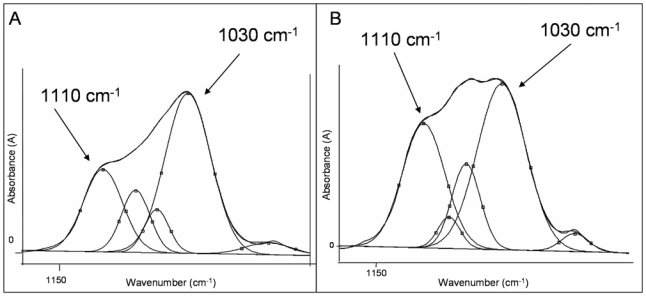
In human bone and at the level of the ν1-ν3PO4 vibration, the 1110 cm−1 peak (non-apatitic domain) is less intense in old bone (A) than in new bone (B). This reflects that mineral maturation of old interstitial bone is greater than that of newly formed bone.
The area ratio 1030/1110 cm−1 was always higher in old interstitial bone than in newly formed bone. A significant difference (p<0.03) was found between cortical and cancellous bone in old bone (Table 1).
Mineral crystallinity index
In human controls, crystallinity index was found to be higher in old interstitial bone than in newly formed bone (FWHM=24.536 in old bone versus 25.078 in new bone, crystallinity index is higher when FWHM is weak). No significant differences were observed between cortical and cancellous bone (Table 1).
Correlation between mineral maturity and crystallinity index in human controls
There was a significant but weak correlation (r′=0.38) between mineral maturity and crystallinity index (Fig. 5D). In human controls, compared with synthetic apatites, the decrease of correlation between mineral maturity and crystallinity index seems mainly due to the presence of a larger amount of non-apatitic domain in human bone than in synthetic apatites. Moreover, it is important to mention that there is a significant difference between cortical and cancellous old bone in mineral maturity but not in crystallinity index.
Observations in skeletal fluorosis
In the two cases analyzed, mineral maturity and crystallinity index showed different patterns. Compared with human controls, mineral crystallinity index was higher and mineral maturity was slightly lower in the fluorosis samples (Table 1).
XRD analysis performed on two pairs of bone samples (fluorotic and normal bone) revealed that the fluorotic bone present overall a higher crystallinity. Analyses of (002) and (310) reflections showed that the (310)-line width was highly decreased in fluorosis compared with normal bone, while (002) reflection was unchanged. FTIRM analysis of the same samples showed that FWHM of 604 cm−1 peak is decreased as well in fluorosis compared with normal bone (in cancellous old bone, FWHM=19.131 in fluorotic bone versus 24.636 in control bone; Fig. 7).
Figure 7.
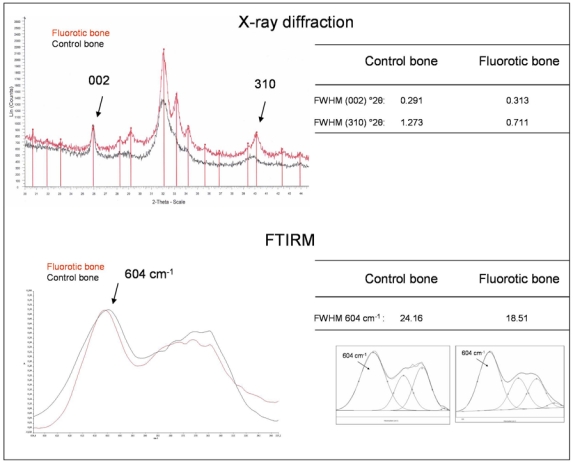
Example of one pair of samples analyzed by X-ray diffraction (XRD) (fluorotic bone in red and control bone in black) and by FTIRM. XRD diagram shows the decrease of the full width at half maximum (FWHM) (°2θ) of the (310) reflection in fluorosis, while (002) reflection is unchanged. Regarding the overall aspect of XRD diagram of the two samples, fluorotic bone is more crystallized than control bone. FTIRM analysis shows a decrease of the FWHM of the 604 cm−1 (peak fitting inserted) of the fluorotic bone, indicating an increase in mineral crystallinity index.
Correlation between mineral maturity and crystallinity index in skeletal fluorosis
No correlation was found between mineral maturity and crystallinity index. Mineral maturity is lower and mineral crystallinity index is higher in fluorotic bone compared with control bone.
Discussion
The main purpose of the present study was to test the hypothesis that, at the BSU level, two distinct parameters were involved in bone mineral quality, i.e., mineral maturity and mineral crystallinity index. Whereas those two parameters often evolve concomitantly, it is a current use to speak about mineral maturity/crystallinity and to join the two parameters into one. By definition, they are not equivalent and correspond to two different entities. Maturity is related to the ratio of apatitic and non-apatitic domains, whereas crystallinity depends on both the perfection of the apatite crystalline domains and their size/strain. We have shown on normal and pathologic bone (fluorotic bone), that the two parameters, mineral maturity and mineral crystallinity index, had to be considered separately. Several attempts have been made to determine mineral maturity and crystallinity index by infrared spectroscopy [10, 11, 27, 28, 32]. Mineral maturity and crystallinity index are often not separated and they are expressed as a single parameter [33, 34]. Several terms have been used to describe bone mineral: crystallinity, maturity, crystal size, crystalline perfection. However, the association of these physicochemical parameters with the characteristics of the crystals: shape, length, width, and ordering of ions, has remained difficult and has sometimes been controversial. During the formation of bone mineral crystals, two main characteristics are noted, reflecting bone mineral characteristics: firstly, mineral maturity, which corresponds to the progressive transformation of immature surface-hydrated domains into a mature and more stable apatite lattice; and secondly, mineral crystallinity index, which corresponds to the size and strains of these apatite domains [35]. It appears essential to separate mineral maturity and crystallinity index, because they do not represent the same mineral characteristics. Our results obtained in human fluorotic bones have required their separation since they corresponded to different mineral entities.
Infrared spectroscopy is a technique providing information especially about covalent bonds of molecules. Chemical groups (e.g. hydroxyl, phosphate, amide) can be identified due to their specific absorption at different wavenumbers. In all the spectroscopic techniques (RMN, Raman, infrared spectroscopies), a modification in the size and strain of crystalline domains involves a broadening and sometimes a distortion of lines, observed on the spectra. It is thus possible to determine a crystallinity index with this technique, like in Raman spectroscopy. Ionic substitutions occurring within apatite domains especially influence the environment of vibrating units and result in broadening of the specific lines related to these crystalline domains. On the contrary, substitutions in the hydrated layer have little or no influence on the crystalline apatite lattice. Consequently, infrared spectroscopy is sensitive to both the chemical environment of molecules, allowing the distinction of surface hydrated layer and crystalline apatite domains and, when focusing on specific apatite lines, the crystallinity of these domains.
During mineral maturation, the hydrated layer decreases while the stable apatite domains grow, corresponding to the evolution of non-apatitic environments of mineral ions into apatitic environments detected by FTIR. This hydrated layer, different from a hydration layer (Stern double layer), corresponds to the mode of formation of apatite crystals in physiologic conditions. The existence of these two domains, a hydrated layer and an apatite core in biomimetic nanocrystals, has been recently confirmed by solid-state NMR [36]. The hydrated surface layer contains loosely bound ions, which are easily exchangeable, and determine the surface properties of the nanocrystalline apatites [1, 3]. In bone, those loosely bound ions can be also exchanged with charged groups of proteins present in collagen and non-collagenous proteins. Several studies have shown the role of charged proteins on mineralization [37–39]. Recently, Dziak and Akkus have shown that some polyelectrolytic peptides could affect the quality of mineral crystals in vitro [40]. In infrared spectroscopy, the ν1ν3PO4 domain presents intense bands generated by phosphate ions, the most intense being the 1030 cm−1 peak, which has been assigned to one component of the triply degenerated ν3PO4 antisymmetric stretching, vibrational mode of the PO4 groups in stoichiometric apatite [13]. The 1020 cm−1 line has been associated with non-stoichiometric poorly crystalline apatite and is present at the early stage of mineralization [28]. Those two peaks have been first identified by Rey et al with the technique of Fourier Self-Deconvolution [13]. Later, Boskey’s group has used the ratio of different peaks in this domain (among 11 peaks) based on second derivative treatment of the spectra of bone [10]. However, in our opinion, the use of a curve-fitting process involving 11 underlying bands to fit the ν1ν3PO4 vibration does not appear reproducible and stable enough, and may lead to different solutions, even if numerous bands improve the criteria of convergence between the computed and the raw spectra. In addition, the assignment and meaning of all these bands is far from being completed. In earlier studies, Pleshko et al used a maximum of six underlying components with the same technique of curve-fitting [11]. Later, Boskey et al have used the correlations established between FTIR and the XRD analyses on synthetic and biologic samples to establish an index mineral maturity/crystallinity, using the 1030/1020 area ratio [10]. This ratio has become a standard largely used in the literature, especially by this group. This ratio increases with the maturation and the crystal size in synthetic samples or normal biologic samples and we agree about this observation. However, according to our interpretations, the ratio 1030/1020 area ratio corresponds to the ratio of apatitic over non-apatitic phosphate and therefore to a mineral maturity index rather than a crystallinity index. The good correlation between FTIR maturity index and the crystallinity provided by XRD does not mean that these two parameters measure the same characteristics of the mineral, but simply means that these characteristics evolve concomitantly in the samples analyzed. It would be erroneous to believe that an increase in the 1030/1020 ratio is necessarily always related to an increase of crystal size. An increase of the 1030/1020 ratio means that there is an increase of the mineral maturity, i.e., a more important proportion of apatitic domain compared with non-apatitic surface domain, meaning the bone is older. But it does not means that the crystal size has necessarily increased, as it has been suggested in some studies [7]. This suggestion is based on the assumption that these parameters are always linked by the same phenomenologic correlation that cannot be ascertained for all bone mineral formation, even if we recognize that, sometimes, the maturation and crystallinity are correlated. For example, if bone mineral contains a substance modifying the crystallinity (for example due to an anti-osteoporotic treatment or a pathology), the mineral maturity can be normal but the crystallinity index increased, simply due to its presence in bone mineral. It does not suggest that the crystals are larger or more mature. From then on, there has been much confusion between the two characteristics: mineral maturity and crystallinity index. In our experiments, the 1030/1110 area ratio was chosen because the 1020 cm−1 line is not clearly resolved on raw spectra and thus is not identifiable compared with the 1110 cm−1 line. Exactly like 1020 cm−1, the 1110 cm−1 peak is present in freshly precipitated apatite and is assigned to the ν3PO4 antisymmetric stretch or HPO4 stretch in poorly crystalline apatite [13, 30, 31]. It disappeared progressively during maturation in biologic and synthetic samples [41]. During the conversion of amorphous calcium phosphate into hydroxyapatite, four peaks (1020, 1038 and 1112 and 1127 cm−1) can be attributable to non-stoichiometry and/or the presence of acid phosphate-containing species [31]. In young bone mineral, the 1110 cm−1 peak is predominant [13] and better resolved compared with the 1020 cm−1 peak. Our results show that the 1110 cm−1 peak is less intense in synthetic apatites than in newly formed BSUs. However, in both synthetic apatites and human bone, the ratio 1030/1110 increased with mineral maturation. Moreover, invariably in human adult controls, old interstitial bone was significantly more mature than newly formed bone. This is the reason why we have chosen the 1110 cm−1 rather the 1020 cm−1 peak. We have also performed on bone control samples the correlation between the 1030/1020 cm−1 and the 1030/1110 cm−1 ratio, using the intensity of raw spectra and not the area ratio (peak curve-fitting; it is important to emphasize once more that the 1020 cm−1 was not clearly identifiable on spectra.). The two ratios are significantly correlated and thus, they show a similar evolution (r′=0.86, p<0.0001).
An important characteristic of bone mineral is its ability to accept ionic substitutions and vacancies. This can affect several parameters like crystallinity of the apatite domains, physicochemical functions, morphology, and crystal size. ν4PO4 vibration has been extensively used to analyze the crystallinity index of the apatite domains in vibrational macroscopic spectroscopy [9, 12, 26, 41], but is often inaccessible for microscopic infrared study, particularly in FTIRI (due to the limit of detection of the cut-off of the detector used in a conventional IR microscope). The use of a wide-band detector allowed access to this part of the infrared spectrum. Crystallinity of the apatite domains can be assessed by several methods (XRD, WAXS, SAXS, vibrational spectroscopy [FTIR and Raman], NMR spectroscopy) [42]. Several parameters have been used to assess crystallinity index by infrared spectroscopy methods on ν1ν3PO4 [11] or ν4PO4 [9, 28]. Several indicators of mineral crystallinity have been proposed. Termine and Posner first used the resolution ratio of the ν4PO4 band, i.e. the splitting factor [28]. Several studies have used similar ratios to describe crystallinity [26, 27]. Bands in the ν4PO4 arise from the antisymmetric P–O bending modes of the phosphate groups. The FWHM of the ν1PO4 line has been commonly used in Raman spectroscopy to represent the apatite crystallinity [43–45]. Increasing crystallinity indicates that a greater proportion of crystals have larger size and/or stoichiometrically more perfect lattice with fewer substitutions [40]. However, infrared spectroscopy is not able to dissociate the influence of crystallite size from the lattice imperfections. We have chosen to analyze ν4PO4 to measure the crystallinity index because it presented well-resolved peaks assigned with certitude to apatite lattice, and it has already been proposed to serve as an IR crystallinity index [9, 27, 28]. Among the different possibilities, we chose the well-resolved 604 cm−1 line. To confirm the robustness of using the FWHM of the 604 cm−1 line (inversely proportional to crystallinity index; the narrower the peak is, the higher the crystallinity index), we have compared, on the same samples, the results obtained with the measurement of another crystallinity index published by Shemesh [27], which is a derived ratio of the splitting factor used by Termine and Posner [28]. There is an excellent and significant correlation between the two crystallinity indexes measured differently on the same phosphate vibration. Therefore, the use of the FWHM, as in the way it is often used in Raman spectroscopy, gives a very good indication of the state of crystallinity of the sample. We chose the crystallinity index of the apatite domains based only on FWHM of the 604 cm−1 line (inversely correlated to crystallinity index), clearly assigned to apatite phosphate groups. This crystallinity index seems more reliable than the resolution factors proposed by different authors, which may depend also on the position variations of the two main apatite phosphate peaks and on the possible presence of underlying non-apatitic phosphate absorption bands. Our results on the 604 cm−1 peak showed that in the mineral of newly formed bone, apatite domains are invariably less well crystallized than in old interstitial bone. This involves a better organization of mineral ions in the apatite lattice in old bone than in newly formed bone. A low crystallinity can be due to the fact that the apatite domains are small and poorly organized in nascent crystals of young bone, but it could also occur as the result of the accelerated remodeling. Another peak (563 cm−1) in the ν4PO4 vibration, not described in this study, is also interesting because it probably contains a contribution of non-apatitic domains [9, 12].
The strong correlation between mineral maturity and crystallinity index in synthetic apatites suggests that these two parameters are temporally linked, and this is in agreement with previous results showing that the area ratio of 1030/1020 cm−1 lines (apatitic PO4/non-apatitic PO4) are well correlated with the crystallinity measured by XRD [32, 34]. Our results are in adequation with those results, showing that mineral maturity evolves generally in parallel with crystallinity index in a normal bone. However, the chemistry and development of biologic apatite is very different compared with synthetically grown apatite. Mineral maturity represents a stage of maturation, whereas crystallinity is involved in the organization of the apatite lattice. Thus in physiologic conditions, it makes sense that mineral maturity and crystallinity index evolve similarly, i.e., that when apatitic environments become more important than non-apatitic environments, apatite domains have grown and have become more perfect. In controls, mineral maturity in old bone was significantly higher in cortical than in cancellous bone, but crystallinity index was not. This suggests that the two parameters evolve differently. A recent study performed in our laboratory by Bala et al. [46] shows that in ewes (an animal model with a remodeling activity close to humans), the kinetics of evolution of the two parameters is different. Mineral maturity is more rapidly completed than crystallinity index. This could suggest that mineral maturity occurs rapidly to reach a certain stability of the crystal, and when this is attained, there is a progressive increase in the crystallinity index with a slow increase in size. This observation reinforces the fact that it is important to separate mineral maturity from crystallinity index. However, it is possible to have a disruption of these parallel evolutions, in certain conditions, for example when an ionic substitution occurs in apatite lattice, In a case of ionic substitution introducing distortions in the apatite lattice, crystallinity index is modified. However, maturity linked to the dynamic of bone remodeling is different from the static effect linked to the state of the mineral, i.e., crystallinity (size/perfection). It is important to emphasize that the correlation between the two parameters with X-ray methods has led to the simultaneous expression of maturity/crystallinity. To verify the hypothesis that incorporation in the apatite lattice of foreign ions could produce a change in crystallinity index without modifications of mineral maturity, these two parameters were measured in samples from patients with skeletal fluorosis. In the latter, the crystallinity index is increased, due to the substitution of OH− by F− ions, which are easily incorporated into the apatite lattice due to their small ionic radius (rF−=1.33 Å, rOH−=1.45 Å), producing a smaller unit cell volume [47]. This is in agreement with other studies showing that the increase in crystallinity was related to the F− content in shark dentine [8], in enamel [29] and in bone mineral [48–50]. However, mineral maturity is slightly decreased. This decrease indicates the presence of young mineral deposition due to the stimulation of osteoblastic activity, enhanced by fluoride [51]. Thus, in spite of the fact that the great order of ions in the apatite lattice in skeletal fluorosis explains the increase in crystallinity index, the mineral maturity is decreased. Although mineral maturity and crystallinity index were not distinguished previously [7, 10, 33, 34, 52], those results emphasize for the first time that the two parameters have to be measured separately.
Separate measurements of mineral maturity and crystallinity index by FTIRM suggested different implications of these parameters in bone strength. Bone mineral crystals are extremely small, inducing a large specific surface of bone crystals, and contributing to an increased quantity of electrostatic bonds between mineral and collagen matrix. Mechanically, the highly ordered location and orientation of very small crystals within the collagen fibrils not only contribute to the rigidity and strength of the bone substance, but their small size also permits an acceptable range of flexibility without fracture or disruption of the bone substance [35, 53]. In fluorosis, bone is very fragile [54–57]. A negative correlation was found between crystal width and fracture stress of femur in rabbits and rats treated with fluoride [55]. The high crystallinity index in fluorosis suggested organization of the crystals, and an increase of the size of the crystals. However, the presence of large crystals decreases the surface area with collagen fibrils and, therefore, does not contribute to mechanical strength [54, 58]. Thus, the bone fragility observed with fluoride, and the modifications in bone mineral shown in the present study in fluorosis, suggest that mineral crystallinity index and mineral maturity could independently influence bone strength. Recently, the importance of mineral crystallinity, investigated by Raman spectroscopy, on mechanical properties of human bone has been demonstrated. Crystallinity alone explains 7–48% of the variation on monotonic mechanical properties, and also 11–63% of the variation of the fatigue properties [59]. It must be also mentioned that the crystal orientation plays an important role in biomechanical properties [20, 60]. A recent study on slices of bovine femur has shown, by XRD analysis, that the deformation of crystals induced by tensile loading was different according to their degree of orientation, and the deformation behavior of mineral crystals depended on structural anisotropy [61].
Anti-osteoporotic treatments act either by a stimulation of bone formation (teriparatide) or by a decrease of bone resorption (bisphosphonates), or by both mechanisms. Consequently, these treatments can influence mineral maturity, with a lowest mineral maturity with anabolic treatments, and conversely a highest mineral maturity with antiresorptive treatments. It has been shown that 3 and 5 years of treatment with risedronate preserved the mineral maturity/crystallinity, whereas after 3 years of placebo, there were significant changes (continued maturation) in the bone matrix, suggesting that risedronate arrested the tissue aging encountered in osteoporosis [62]. Crystallinity can also be influenced by the mineralization rate, and this has been shown in osteoporosis where the increase in bone remodeling increased the mineral maturity/crystallinity parameter [33]. These data underscore the simultaneous measurement of several parameters involved in bone mineral traits for a better understanding of the mechanisms of action of the various treatments of osteoporosis.
Three mains limitations appeared in our study. Firstly, it should be emphasized that this study analyzes intrinsic characteristics of bone mineral, and not the remodeling activity. Thus, measurements did not take into account the relative amount of bone, but were done at the BSU level as a function of the age of bone. Secondly, infrared spectroscopy was aimed at the mid-range order compared with XRD, which gave long-range order information and directly addressed the distances and regularity of the arrangements of atoms/molecules in a crystal lattice. Thus, infrared spectroscopy was less sensitive to modification of the long-range order, while XRD allowed it. Thirdly, the small number of samples reduced the ability to detect differences with age, as shown by Hanschin [63]. They showed by XRD that the most important variations appeared within the first 30 years, with a distinct reduction in the peak width of the (002) and (310) reflection.
In conclusion, we have demonstrated that mineral maturity and crystallinity index are two different parameters that do not refer to the same domain of crystals. These two parameters need to be distinguished in later studies on bone mineral. They will then be used to analyze modifications of bone quality at the crystal level in bone samples from osteoporotic patients treated, or not, for a better understanding of the mechanisms of bone fragility.
Acknowledgments
The authors express their gratitude to Ruben Vera (Centre de Diffractométrie Henri Longchambon, Université de Lyon, France) for performing the XRD analyses, and to Monique Arlot for her help in statistical analysis. This work was supported in part by an unrestricted educational grant from Eli Lilly to INSERM.
Footnotes
The authors have no conflict of interest.
References
- 1.Green J. The physicochemical structure of bone: cellular and noncellular elements. Miner Electrolyte Metab. 1994;20:7–15. [PubMed] [Google Scholar]
- 2.Cazalbou S. PhD thesis. University of toulouse; france: 2000. [Google Scholar]
- 3.Cazalbou S, Combes C, Eichert D, Rey C, Glimcher MJ. Poorly crystalline apatites: evolution and maturation in vitro and in vivo. J Bone Miner Metab. 2004;22:310–317. doi: 10.1007/s00774-004-0488-0. [DOI] [PubMed] [Google Scholar]
- 4.Barry AB, Baig AA, Miller SC, Higuchi WI. Effect of age on rat bone solubility and crystallinity. Calcif Tissue Int. 2002;71:167–171. doi: 10.1007/s00223-001-1071-5. [DOI] [PubMed] [Google Scholar]
- 5.Ager JW, Nalla RK, Breeden KL, Ritchie RO. Deep-ultraviolet Raman spectroscopy study of the effect of aging on human cortical bone. J Biomed Opt. 2005;10:034012. doi: 10.1117/1.1924668. [DOI] [PubMed] [Google Scholar]
- 6.Akkus O, Polyakova-Akkus A, Adar F, Schaffler MB. Aging of microstructural compartments in human compact bone. J Bone Miner Res. 2003;18:1012–1019. doi: 10.1359/jbmr.2003.18.6.1012. [DOI] [PubMed] [Google Scholar]
- 7.Boskey AL, DiCarlo E, Paschalis E, West P, Mendelsohn R. Comparison of mineral quality and quantity in iliac crest biopsies from high- and low-turnover osteoporosis: an FT-IR microspectroscopic investigation. Osteoporos Int. 2005;16:2031–2038. doi: 10.1007/s00198-005-1992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Legeros RZ. Apatites in biological systems. ProgCrystal Growth Charact. 1981;4:1–45. [Google Scholar]
- 9.Miller LM, Vairavamurthy V, Chance MR, Mendelsohn R, Paschalis EP, Betts F, Boskey AL. In situ analysis of mineral content and crystallinity in bone using infrared microspectroscopy of the nu(4) PO(4)(3-) vibration. Biochim Biophys Acta. 2001;1527:11–19. doi: 10.1016/s0304-4165(01)00093-9. [DOI] [PubMed] [Google Scholar]
- 10.Paschalis EP, DiCarlo E, Betts F, Sherman P, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of human osteonal bone. Calcif Tissue Int. 1996;59:480–487. doi: 10.1007/BF00369214. [DOI] [PubMed] [Google Scholar]
- 11.Pleshko N, Boskey A, Mendelsohn R. Novel infrared spectroscopic method for the determination of crystallinity of hydroxyapatite minerals. Biophys J. 1991;60:786–793. doi: 10.1016/S0006-3495(91)82113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rey C, Shimizu M, Collins B, Glimcher MJ. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ions in the early deposits of a solid phase of calcium-phosphate in bone and enamel, and their evolution with age. I: Investigations in the upsilon 4 PO4 domain. Calcif Tissue Int. 1990;46:384–394. doi: 10.1007/BF02554969. [DOI] [PubMed] [Google Scholar]
- 13.Rey C, Shimizu M, Collins B, Glimcher MJ. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ion in the early deposits of a solid phase of calcium phosphate in bone and enamel and their evolution with age: 2. Investigations in the nu3PO4 domain. Calcif Tissue Int. 1991;49:383–388. doi: 10.1007/BF02555847. [DOI] [PubMed] [Google Scholar]
- 14.Akkus O, Adar F, Schaffler MB. Age-related changes in physicochemical properties of mineral crystals are related to impaired mechanical function of cortical bone. Bone. 2004;34:443–453. doi: 10.1016/j.bone.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Morris MD, Finney WF, Rajachar RM, Kohn DH. Bone tissue ultrastructural response to elastic deformation probed by Raman spectroscopy. Faraday Discuss. 2004;126:159–168. doi: 10.1039/b304905a. discussion 169–183. [DOI] [PubMed] [Google Scholar]
- 16.Tarnowski CP, Ignelzi MA, Jr, Wang W, Taboas JM, Goldstein SA, Morris MD. Earliest mineral and matrix changes in force-induced musculoskeletal disease as revealed by Raman microspectroscopic imaging. J Bone Miner Res. 2004;19:64–71. doi: 10.1359/JBMR.0301201. [DOI] [PubMed] [Google Scholar]
- 17.Carden A, Rajachar RM, Morris MD, Kohn DH. Ultrastructural changes accompanying the mechanical deformation of bone tissue: a Raman imaging study. Calcif Tissue Int. 2003;72:166–175. doi: 10.1007/s00223-002-1039-0. [DOI] [PubMed] [Google Scholar]
- 18.Miller LM, Little W, Schirmer A, Sheik F, Busa B, Judex S. Accretion of bone quantity and quality in the developing mouse skeleton. J Bone Miner Res. 2007;22:1037–1045. doi: 10.1359/jbmr.070402. [DOI] [PubMed] [Google Scholar]
- 19.Carden A, Morris MD. Application of vibrational spectroscopy to the study of mineralized tissues (review) J Biomed Opt. 2000;5:259–268. doi: 10.1117/1.429994. [DOI] [PubMed] [Google Scholar]
- 20.Fratzl P, Gupta HS, Paschalis EP, Roschger P. Structure and mechanical quality of the mineral quality of the collagen-mineral nano-composite in bone. J Mater Chem. 2004;14:2115–2123. [Google Scholar]
- 21.Paschalis EP, Glass EV, Donley DW, Eriksen EF. Bone mineral and collagen quality in iliac crest biopsies of patients given teriparatide: new results from the fracture prevention trial. J Clin Endocrinol Metab. 2005;90:4644–4649. doi: 10.1210/jc.2004-2489. [DOI] [PubMed] [Google Scholar]
- 22.Miller LM, Novatt JT, Hamerman D, Carlson CS. Alterations in mineral composition observed in osteoarthritic joints of cynomolgus monkeys. Bone. 2004;35:498–506. doi: 10.1016/j.bone.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 23.Huang RY, Miller LM, Carlson CS, Chance MR. In situ chemistry of osteoporosis revealed by synchrotron infrared microspectroscopy. Bone. 2003;33:514–521. doi: 10.1016/s8756-3282(03)00233-3. [DOI] [PubMed] [Google Scholar]
- 24.Rey C, Hina A, Tofighi A, Glimcher MJ. Maturation of poorly crystalline apatites: chemical and structural aspects in vivo and in vitro. Cells and Mater. 1995;5:345–356. [Google Scholar]
- 25.Gee A, Dietz VR. Pyrophosphate formation upon ignition of precipitated basic calcium phosphate. J Am Chem Soc. 1955;77:2961–2965. [Google Scholar]
- 26.Pucéat E, Reynard B, Lécuyer C. Can crystallinity be used to determine the degree of chemical alteration of biogenic apatites? Chemical Geology. 2004;205:83–97. [Google Scholar]
- 27.Shemesh A. Crystallinity and diagenesis of sedimentary apatites. Geochimica et Cosmochimica Acta. 1990;54:2433–2438. [Google Scholar]
- 28.Termine JD, Posner AS. Infra-red determinaion of the percentage of crystallinity in apatitic calcium phosphates. Nature. 1966;211:268–270. doi: 10.1038/211268a0. [DOI] [PubMed] [Google Scholar]
- 29.Frazier PD, Little MF, Casciani FS. X-ray diffraction analysis of human enamel containing different amounts of fluoride. Arch Oral Biol. 1967;12:35–42. doi: 10.1016/0003-9969(67)90139-2. [DOI] [PubMed] [Google Scholar]
- 30.Bohic S, Heymann D, Pouezat JA, Gauthier O, Daculsi G. Transmission FT-IR microspectroscopy of mineral phases in calcified tissues. C R Acad Sci III. 1998;321:865–876. doi: 10.1016/s0764-4469(99)80027-4. [DOI] [PubMed] [Google Scholar]
- 31.Gadaleta SJ, Paschalis EP, Betts F, Mendelsohn R, Boskey AL. Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif Tissue Int. 1996;58:9–16. doi: 10.1007/BF02509540. [DOI] [PubMed] [Google Scholar]
- 32.Camacho NP, Rinnerthaler S, Paschalis EP, Mendelsohn R, Boskey AL, Fratzl P. Complementary information on bone ultrastructure from scanning small angle X-ray scattering and Fourier-transform infrared microspectroscopy. Bone. 1999;25:287–293. doi: 10.1016/s8756-3282(99)00165-9. [DOI] [PubMed] [Google Scholar]
- 33.Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of human iliac crest biopsies from untreated osteoporotic bone. Calcif Tissue Int. 1997;61:487–492. doi: 10.1007/s002239900372. [DOI] [PubMed] [Google Scholar]
- 34.Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of normal human cortical and trabecular bone. Calcif Tissue Int. 1997;61:480–486. doi: 10.1007/s002239900371. [DOI] [PubMed] [Google Scholar]
- 35.Glimcher MG. The nature of the mineral phase in bone: Biological and clinical implications. Metabolic bone disease. 1998;Chap 2:23–50. [Google Scholar]
- 36.Jager C, Welzel T, Meyer-Zaika W, Epple M. A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn Reson Chem. 2006;44:573–580. doi: 10.1002/mrc.1774. [DOI] [PubMed] [Google Scholar]
- 37.Boskey A, Maresca M, Appel J. The effects of noncollagenous matrix proteins on hydroxyapatite formation and proliferation in a collagen gel system. Connect Tissue Res. 1989;21:171–176. doi: 10.3109/03008208909050007. discussion 177–178. [DOI] [PubMed] [Google Scholar]
- 38.Boskey AL. Noncollagenous matrix proteins and their role in mineralization. Bone Miner. 1989;6:111–123. doi: 10.1016/0169-6009(89)90044-5. [DOI] [PubMed] [Google Scholar]
- 39.Boskey AL, Gadaleta S, Gundberg C, Doty SB, Ducy P, Karsenty G. Fourier transform infrared microspectroscopic analysis of bones of osteocalcin-deficient mice provides insight into the function of osteocalcin. Bone. 1998;23:187–196. doi: 10.1016/s8756-3282(98)00092-1. [DOI] [PubMed] [Google Scholar]
- 40.Dziak KL, Akkus O. Effects of polyelectrolytic peptides on the quality of mineral crystals grown in vitro. J Bone Miner Metab. 2008;26:569–575. doi: 10.1007/s00774-008-0869-x. [DOI] [PubMed] [Google Scholar]
- 41.Rey C, Beshah K, Griffin R, Glimcher MJ. Structural studies of the mineral phase of calcifying cartilage. J Bone Miner Res. 1991;6:515–525. doi: 10.1002/jbmr.5650060514. [DOI] [PubMed] [Google Scholar]
- 42.Runt J, Kanchanasopa M. Crystallinity determination. [Google Scholar]
- 43.Ferrari AC, Robertson J. Raman spectroscopy of amourphous nanostrucured, diamnd-like carbon, and nanodiamnd. Phil Trans R Soc Lond. 2004;362:2477–2512. doi: 10.1098/rsta.2004.1452. [DOI] [PubMed] [Google Scholar]
- 44.Fitzer EGE, Rozploch F, Steinert D. Application of laser-Raman spectroscopy for characterization of carbon fibres. High Temp high pressures. 1987;19:537–544. [Google Scholar]
- 45.Nasdala L, Pidgeon RT, Wolf D. Heterogeneous metamictization of zircon on a microscale. Geochim Cosmochim Acta. 1996;60:1091–1097. [Google Scholar]
- 46.Bala Y, Farlay D, Delmas PD, Meunier PJ, Boivin G. Time sequence of secondary mineralization and microhardness in cortical and cancellous bone from ewes. Bone. 2009 doi: 10.1016/j.bone.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 47.Grynpas MD. Fluoride effects on bone crystals. J Bone Miner Res. 1990;5(Suppl 1):S169–175. doi: 10.1002/jbmr.5650051362. [DOI] [PubMed] [Google Scholar]
- 48.Posner AS, Eanes ED, Harper RA, Zipkin I. X-Ray Diffraction Analysis of the Effect of Fluoride on Human Bone Apatite. Arch Oral Biol. 1963;168:549–570. doi: 10.1016/0003-9969(63)90071-2. [DOI] [PubMed] [Google Scholar]
- 49.Bang S, Boivin G, Gerster JC, Baud CA. Distribution of fluoride in calcified cartilage of a fluoride-treated osteoporotic patient. Bone. 1985;6:207–210. doi: 10.1016/8756-3282(85)90002-x. [DOI] [PubMed] [Google Scholar]
- 50.Fratzl P, Roschger P, Eschberger J, Abendroth B, Klaushofer K. Abnormal bone mineralization after fluoride treatment in osteoporosis: a small-angle x-ray-scattering study. J Bone Miner Res. 1994;9:1541–1549. doi: 10.1002/jbmr.5650091006. [DOI] [PubMed] [Google Scholar]
- 51.Boivin G, Chavassieux P, Chapuy MC, Baud CA, Meunier PJ. Skeletal fluorosis: histomorphometric analysis of bone changes and bone fluoride content in 29 patients. Bone. 1989;10:89–99. doi: 10.1016/8756-3282(89)90004-5. [DOI] [PubMed] [Google Scholar]
- 52.Faibish D, Ott SM, Boskey AL. Mineral changes in osteoporosis: a review. Clin Orthop Relat Res. 2006;443:28–38. doi: 10.1097/01.blo.0000200241.14684.4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Landis WJ. The strength of a calcified tissue depends in part on the molecular structure and organization of its constituent mineral crystals in their organic matrix. Bone. 1995;16:533–544. doi: 10.1016/8756-3282(95)00076-p. [DOI] [PubMed] [Google Scholar]
- 54.Chachra D, Turner CH, Dunipace AJ, Grynpas MD. The effect of fluoride treatment on bone mineral in rabbits. Calcif Tissue Int. 1999;64:345–351. doi: 10.1007/s002239900630. [DOI] [PubMed] [Google Scholar]
- 55.Turner CH, Garetto LP, Dunipace AJ, Zhang W, Wilson ME, Grynpas MD, Chachra D, McClintock R, Peacock M, Stookey GK. Fluoride treatment increased serum IGF-1, bone turnover, and bone mass, but not bone strength, in rabbits. Calcif Tissue Int. 1997;61:77–83. doi: 10.1007/s002239900299. [DOI] [PubMed] [Google Scholar]
- 56.Turner CH, Hasegawa K, Zhang W, Wilson M, Li Y, Dunipace AJ. Fluoride reduces bone strength in older rats. J Dent Res. 1995;74:1475–1481. doi: 10.1177/00220345950740080701. [DOI] [PubMed] [Google Scholar]
- 57.Turner CH, Boivin G, Meunier PJ. A mathematical model for fluoride uptake by the skeleton. Calcif Tissue Int. 1993;52:130–138. doi: 10.1007/BF00308322. [DOI] [PubMed] [Google Scholar]
- 58.Li Y, Liang C, Slemenda CW, Ji R, Sun S, Cao J, Emsley CL, Ma F, Wu Y, Ying P, Zhang Y, Gao S, Zhang W, Katz BP, Niu S, Cao S, Johnston CC., Jr Effect of long-term exposure to fluoride in drinking water on risks of bone fractures. J Bone Miner Res. 2001;16:932–939. doi: 10.1359/jbmr.2001.16.5.932. [DOI] [PubMed] [Google Scholar]
- 59.Yerramshetty JS, Akkus O. The associations between mineral crystallinity and the mechanical properties of human cortical bone. Bone. 2008;42:476–482. doi: 10.1016/j.bone.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Tadano S, Giri B, Sato T, Fujisaki K, Todoh M. Estimating nanoscale deformation in bone by X-ray diffraction imaging method. J Biomech. 2008;41:945–952. doi: 10.1016/j.jbiomech.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Giri B, Tadano S, Fujisaki K, Sasaki N. Deformation of mineral crystals in cortical bone depending on structural anisotropy. Bone. 2009;44:1111–1120. doi: 10.1016/j.bone.2009.01.394. [DOI] [PubMed] [Google Scholar]
- 62.Durchschlag E, Paschalis EP, Zoehrer R, Roschger P, Fratzl P, Recker R, Phipps R, Klaushofer K. Bone material properties in trabecular bone from human iliac crest biopsies after 3- and 5-year treatment with risedronate. J Bone Miner Res. 2006;21:1581–1590. doi: 10.1359/jbmr.060701. [DOI] [PubMed] [Google Scholar]
- 63.Hanschin RG, Stern WB. X-ray diffraction studies on the lattice perfection of human bone apatite (Crista iliaca) Bone. 1995;16:355S–363S. [PubMed] [Google Scholar]