

Abstract
Background
GM1 gangliosidosis (GM1) is an autosomal recessive lysosomal storage disease caused by deficiency of acid beta-galactosidase (GLB1; EC3.2.1.23). Here, we identify three novel mutations in the GLB1 gene from two Han Chinese patients with GM1 that appear correlated with clinical phenotype.
Methods
One of the two Han Chinese patients with GM1 presented with the juvenile form, and the other with the infantile form with cardiac involvement. Sequencing of the entire GLB1 gene revealed three novel mutations (p.H102 D, p.G494V, c.495_497delTCT), which were absent in 94 normal controls. Transient expression of cDNA encoding these variants was performed in COS-1 cells to evaluate β-galactosidase activities.
Results
The first case (patient 1) with the juvenile form contained two missense mutations, p.H102 D and p.A301V. Patient 2 diagnosed with the infantile form of the disease with cardiac involvement was compound heterozygous for p.G494V and c.495_497delTCT mutations. All mutant beta-galactosidases exhibited significantly reduced activity (12%, 0%, 0%, and 0% for p.H102 D, p.A301V, p.G494V, and c.495_497delTCT), compared with the wild-type beta-galactosidase cDNA clone. The mutations identified in patient 2 with cardiomyopathy were localized in the GLB1 gene region common to both lysosomal beta-galactosidase and elastin binding protein (EBP), and caused a deletion in the elastin-binding domain of EBP.
Conclusions
All four mutations identified in Han Chinese patients induce significant suppression of β-galactosidase activity, correlating with severity of disease and presence of cardiomyopathy.
Introduction
Human β-galactosidase (E.C.3.2.1.23; MIM# 230500) is an lysosomal enzyme that removes β-ketosidically linked galactose residues from glycoproteins, sphingolipids, and keratin sulfate [1]. Deficiency of acid β-galactosidase leads to two metabolic storage diseases, specifically, GM1 gangliosidosis (GM1) and Morquio B disease (MBD, mucopolysaccharidosis type IVB, MPS IVB), inherited as autosomal recessive traits. In GM1 gangliosidosis, absence or suppression of β-galactosidase activity causes excessive accumulation of GM1 ganglioside in neuronal tissue while β-linked galactose-terminal oligosaccharides arising from the lysosomal digestion of glycoproteins are stored in visceral organs and excreted in urine. Three major clinical phenotypes have been classified to date: infantile (type I), late infantile/juvenile (type II), and adult (type III) forms [1]. The most severe infantile form with onset between birth and 6 months results in rapidly progressive central nervous system (CNS) degeneration, visceromegaly, cherry-red spots, skeletal abnormalities, and death usually occurring within the first two years of life. Cardiomyopathy is an atypical clinical feature in some Caucasian patients with infantile GM1 gangliosidosis. The residual β-galactosidase activity in fibroblasts from patients is less than 1.3%, compared to the physiological level. However, the juvenile and adult forms of GM1 gangliosidosis display a less severe course, later age of onset, and higher β-galactosidase activity, varying from 0.3 to 4.8% of the normal level in the juvenile form and ~9% in the adult form [1,2].
In contrast to GM1 gangliosidosis, Morquio B disease (MBD) is characterized by severe skeletal dysplasia without CNS involvement. MBD is also associated with deficiency of acid β-galactosidase, but the metabolic storage substance in this case is keratin sulfate, a glycosaminoglycan accumulating in the cornea and skeletal tissue and secreted in urine, and not GM1 ganglioside in the brain [1].
The human β-galactosidase gene (GLB1) located on chromosome 3p21.33 [3] contains 16 exons spanning approximately 62.5 kb [3,4]. The GLB1 gene encodes two alternatively spliced products, lysosomal β-galactosidase (GLB1) and elastin-binding protein (EBP) [5-7]. The GLB1 protein is synthesized as an N-glycosylated 85 kDa precursor, and processed within lysosomes to a 64 kDa mature enzyme [8-10]. The active enzyme forms complexes with the 54 kDa protective protein/cathepsin A (PPCA) and the 46 kDa neuraminidase (NEU1) in lysosomes. PPCA is essential for intracellular transport and intralysosomal activity/stabilization of both β-galactosidase and NEU1 [11,12]. NEU1 catalyzes hydrolysis of the terminal sialic acids of oligosaccharides, glycoproteins and glycolipids [13,14]. The alternative splice product, EBP, results from deletion of exons 3, 4 and 6, and a frameshift in the translation of exon 5 encoding the unique elastin binding domain of EBP [5,15].
Cardiomyopathy in GM1 gangliosidosis is associated with impaired elastogenesis and EBP defects [16-19]. EBP, a 67 kDa enzymatically inactive variant of β-galactosidase, is not targeted to lysosomes. Earlier studies show that EBP acts as a recycling chaperone that protects tropoelastin from premature degradation and delivers it through an intracellular secretory pathway to the cell surface where tropoelastin is secreted into the extracellular matrix and assembled [6,20-22]. Tropoelastin is released from EBP following a conformational change that occurs after the galactolectin domain of EBP binds to galatosugars protruding from glycoproteins on the microfibrillar scaffold of growing elastic fibers [6,21]. Following tropoelastin release, EBP returns to the endosomes, binds to newly synthesized tropoelastin in the trans-Golgi network, and delivers it to the cell surface [23]. EBP additionally forms complexes with PPCA and NEU1 to facilitate elastic fiber assembly on the cell surface [24]. PPCA appears to act as a protective protein, while NEU1 is required for the release of tropoelastin and the signaling pathway induced by the complex [25,26].
To date, More than 130 mutations have been identified in the GLB1 gene [2,27,28]. In this study, we investigate the GLB1 mutations associated with GM1 gangliosidosis in two Han Chinese patients. Three of the mutations identified in these two patients were novel and have not been reported in patients of other ethnicities. We further evaluate the contribution of individual mutations to the clinical phenotype via site-directed mutagenesis experiments, examination of expression levels in COS-1 cells, and β-galactosidase activity measurements. One of the patients (patient 2) with cardiac involvement presented two mutations likely to affect both lysosomal GLB1 and EBP proteins, including a deletion in the elastin-binding domain of EBP. Our findings suggest a correlation between these mutations and the presence of cardiomyopathy.
Patients and Methods
Patients
The most important clinical features of two GM1 gangliosidosis patients are compared in Table 1. Patient 1, a female, was born after a normal pregnancy. She was the second child of non-consanguineous healthy parents, and the family history was non-contributory. The patient appeared normal at birth. At 6 months of age, abdominal distention, and subsequently, progressive developmental regression, delayed psychomotor development, and recurrent pulmonary infection were observed. At the age of 18 months, the patient displayed coarse facial features, thick skin with Mongolian spots, macroglossia, hepatosplenomegaly, scoliosis, and progressive leukomalacia. MR imaging revealed myelination arrest in the subcortical white matter, and cortical atropy. Enzyme assay of cultured fibroblasts disclosed 2% residual activity compared to the normal level. The patient's clinical condition deteriorated rapidly, and she died of severe emaciation and hypovolemic shock at 2 years, 2 months of age. Based on residual β-galactosidase activity, onset age, disease progression and age at death, the patient was diagnosed with the juvenile form of the disease [1,2].
Table 1.
Clinical and molecular features of two Taiwanese patients with GM1 gangliosidosis
| Patient 1 | Patient 2 | |
|---|---|---|
| Phenotype | Juvenile | Infantile |
| Age of onset | 6 months | Birth |
| Age at diagnosis | 18 months | 4 months |
| Presentation | Developmental regression, delayed psychomotor development, coarse facial features, macroglossia, abdominal distention, and thick skin with Mongolian spots | Developmental regression, hypotonia, bilateral hydrocele, and visceromegaly |
| Eye | Normal | Cherry red spot |
| Heart | Normal | Dilated cardiomyopathy, Hypertrophied left atrium/ventricle, and poor contractility of left ventricle |
| Skeleton | Scoliosis and beaking of the lumbar vertebral bodies | Normal |
| Liver/spleen | Hepatosplenomegaly | Hepatomegaly |
| Nervous system | White matter demyelination and cortical atropy | White matter demyelination, cortical atropy, and seizure |
| Bone marrow | Foamy histiocytes and progressive leukomalacia | Foamy histiocytes |
| Age of death | 2 years, 2 months | 2 years |
| β-galactosidase activity | 2% of control value in fibroblast | < 1% of control value in fibroblast |
| Genetic defect | ||
| Allele 1 | c.304C > G (p.H102 D, inherited from father) | c.495_497delTCT (p.L166del, inherited from mother) |
| Allele 2 | c.902C > T (p.A301V, inherited from mother) | c.1481G > T (p.G494V, inherited from father) |
| Polymorphisms | p.L10P | p.L10P |
Patient 2 presented with the infantile form of the disease, and was the first child of a healthy non-consanguineous couple with no contributory family history. He had developmental retardation, and visceromegaly developed soon after birth. At 3 months of age, the patient presented with macular cherry-red spots and hepatomegaly. Hypotonia and bilateral hydrocele were noted at 4 months of age. The roentgenogram and echocardiogram findings included cardiomegaly, dilated cardiomyopathy, hypertrophied left atrium/ventricle, and poor contractility of the left ventricle. Residual β-galactosidase activity was lower than 1% of the normal level. MR imaging at 3, 12 and 23 months revealed significant myelination arrest with dysmyelination of white matter in the bilateral cerebral hemispheres and atropic change of the brain cortex [29]. The patient died of congestive heart failure and seizure at 2 years of age.
Healthy controls were randomly selected from the Taiwan Han Chinese Cell and Genome Bank [30]. All participating patients and controls were Han Chinese, which is the origin of 98% of the Taiwanese population. The study was approved by the institutional review board and the ethics committee of each institution. Written informed consent was obtained from participants, in accordance with institutional requirements and the Declaration of Helsinki Principles.
Mutation analysis
Genomic DNA was isolated from peripheral blood or fibroblasts using standard techniques. We sequenced all exons of the β-galactosidase gene (GLB1), including the 5' and 3' untranslated regions and intron/exon boundaries. PCR primers were designed using the Primer 3 program http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi. Amplified products from genomic DNA were purified from the agarose gel using QIAEX II (Qiagen, Hilden, Germany), and directly sequenced in both directions using a BigDye Terminator cycle sequencing kit with an ABI Prism 377 DNA Sequencer (Applied Biosystems, Foster City, CA). The GLB1 genomic sequence was derived from GenBank, NT_022517.16. Nucleotide numbering was based on the cDNA reference sequence from GenBank, NM_000404.2, according to the journal guidelines http://www.hgvs.org/mutnomen, with A of the ATG start codon as +1.
Construction of the GLB1 expression plasmid, pGS3
Total RNA was extracted from a normal lymphocyte cell line, and cDNA synthesized using the Advantage RT-for-PCR kit (Clontech, Palo Alto, CA) with an oligo-dT primer. Amplification of wild-type GLB1 gene was performed with the following oligonucleotides: forward, 5'-GTCATGCCGGGGTTCCTGGTTC-3', and reverse, 5'-GTCCCTGAAGGTGGGGCTTTGG-3', using the Expand™ High Fidelity PCR system (Boehringer Mannheim, Indianapolis, IN). The 2.21 kb PCR product was ligated into the TOPO TA cloning kit vector (Invitrogen, Carlsbad, CA), and the nucleotide sequence and orientation of the insert in the resulting plasmid, pTA-GS, verified by direct sequencing. The HindIII-XbaI fragment of pTA-GS was further subcloned into the expression vector, pcDNA3.1 (Invitrogen, Carlsbad, CA), generating the GLB1 expression plasmid, pGS3.
Construction of mutant β-galactosidase expression plasmids with deletion and missense mutations
Site-directed mutagenesis using the recombinant polymerase chain reaction method [31] was performed to introduce the p.His102Asp, c.495_497delTCT, p.Ala301Val, and p.Gly494Val mutations into wild-type GLB1 cDNA. Wild-type pGS3 was employed as the template, and the primers used for site-directed mutagenesis are presented in Table 2. Nucleotide sequences of the resulting mutant GLB1 expression plasmids, pGS3-H102 D, pGS3-c.495_497delTCT, pGS3-A301V and pGS3-G494V, were confirmed for the entire coding region via direct sequencing.
Table 2.
Primers used for site-directed mutagenesis
| Genetic variation | Amino acid change | Exon | Oligonucleotides (5' -> 3') |
|---|---|---|---|
| c.304C > G | p.His102Asp | 3 | F: AACTGGTACTGTCCTGGCCAGGGCTCATGAAAGTTCCAG |
| R: CCTGGCCAGGACAGTACCAGTTTTCTGAGGACGATGATGTGGAATATTTTC | |||
| c.495_497delTCT | p. L166del | 5 | F: ACCACTTGTCCACAGCTGCCAGGTAATCTGGGTCGGAGG |
| R: CTGGCAGCTGTGGACAAGTGGTTGGGAGTCCT---GCCCAAGATGAAGCCTC | |||
| c.902C > T | p.Ala301Val | 8 | F: AAGTATATCATAGAGGGAGGAAGCCACTGCTTCGGTCTTG |
| R: TTCCTCCCTCTATGATATACTTGCCCGTGGGGTGAGTGTGAACTTGTACATG | |||
| c.1481G > T | p.Gly494Val | 15 | F: GTTGATATATGCACCATAGTTCACACGTCCCATGTTCTC |
| R: GAACTATGGTGCATATATCAACGATTTTAAGGTTTTGGTTTCTAACCTG |
1Nucleotide changes are marked with an underline (nucleotide substitution) or a string of hyphens (deletion).
Transient transfection of COS-1 cells
COS-1 cells were cultured in DMEM and 10% fetal bovine serum supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. Cells were grown to 85-90% confluency in each well of a 6-well plate, and separately transfected with 4 μg of pGS3-H102 D, pGS3-c.495_497delTCT, pGS3-A301V, pGS3-G494V and wild-type pGS3. Transfection was performed using the Lipofectamine™ 2000 reagent (Gibco BRL, Grand Island, NY), according to the manufacturer's instructions. After 48 h incubation at 37°C, cells were scraped and washed twice with PBS. Cell pellets were frozen at -70°C until use for protein content determination and assay of enzyme activity.
Enzyme assays
Transfected cells were resuspended in distilled water, and subjected to sonication using a Misonix Ultrasonic processor (XL2020) at a power level of 10 for 15 s in ice. Soluble lysates were collected and protein concentrations determined using the Bio-Rad assay kit (Hercules, CA) with bovine serum albumin as a standard. The GLB1 enzyme assay was performed using the artificial substrate, 4-methylumbelliferyl-β-D-galactopyranoside (Sigma, St. Louis, MO) [32,33]. Briefly, β-galactosidase activity was measured in duplicate by adding 100 μl of enzyme solution to 300 μl of 0.65 mM substrate solution (4-methylumbelliferyl-β-D-galactopyranoside in 0.1 M citrate-phosphate buffer, pH 4.2). After incubation at 37°C for 1 h, all reactions were terminated by adding 1 ml of 0.1 M 2-methylpropan-1-ol, pH 10. Fluorescence was determined with a PerSeptive Biosystems (Cytofluor® series 4000) fluorescence multi-well plate reader (Applied Biosystems, Foster City, CA). A known amount of 4-methylumbelliferone (Sigma, St. Louis, MO) in 0.1 M 2-methylpropan-1-ol, pH 10, was employed as the standard.
Results
Molecular analysis of GM1-gangliosidosis patients
Genomic DNA from two Han Chinese patients with GM1 was amplified with a view to screening all 16 exons and splice junctions of the GLB1 gene via direct sequencing. In total, one deletion, three missense mutations and one polymorphism were identified (Tables 1 and 2). Among these, three mutations (p.H102 D, p.G494V, and c.495_497delTCT) were novel. Patient 1 diagnosed with the juvenile form of GM1 contained two mutations, specifically, c.304C > G in exon 3 (p. H102D) and c. 902C > T in exon 8 (p. A301V)[28] (Fig.1A and 1B), as well as one polymorphism in exon 1, homozygous p.L10P polymorphism (rs.7637099, data not shown). Patient 2 presenting with the infantile form of the disease contained a 3 bp in-frame deletion in exon 5, resulting in deletion of a residue, both in the GLB1 protein (c.495_497delTCT, p. L166del, Fig. 1D) and elastin-binding domain of the EBP protein (c.283_285delTCT, p.S95del, Fig.2). Another nucleotide mutation, c.1481G > T in exon 15, led to the generation of p.G494V (Fig. 1C). Patient 2 was heterozygous for the p.L10P polymorphism (data not shown). These variations in the GLB1 gene were confirmed in parent genomic DNA (Table 1). The p.L10P polymorphism was identified in the 94 normal Han Chinese subjects at an allele frequency of 47.3% (87 of 184 alleles) while the p.His102Asp, c.495_497delTCT (p.Leu166del), p.Ala301Val, and p.Gly494Val variants were absent in the 94 normal Han Chinese subjects. Alignment of the GLB1 sequences from human, mouse, rat, cow, cat, dog, and chicken revealed that p.Leu166, Ala301, and Gly494 are conserved throughout these species. All residues corresponding to human p.His102 among the species examined are basic (histidine, arginine or glutamine) whereas mutant p.Asp102 contains an acidic residue (Fig. 3). Gene variants were further analyzed for functional consequences on GLB1 enzyme activity in COS-1 cells.
Figure 1.

Nucleotide sequences of the neighboring regions of the mutations in the GLB1 gene of two GM1 gangliosidosis patients. The mutant sequencing diagrams are shown. The position of each mutation is marked with an arrow (panels A-D). Wild-type and mutant nucleotide and amino acid sequences are presented using A of the ATG start codon of GLB1cDNA as position +1.
Figure 2.
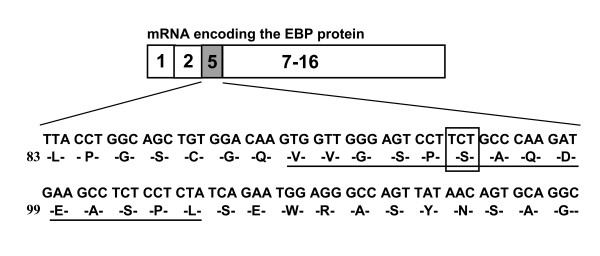
Schematic representation of alternatively spliced mRNA of the GLB1 gene encoding EBP. The unique 32 amino acid region encoded by a frameshift in exon 5 contains an elastin/laminin-binding domain (underlined). The 3 bp in-frame deletion resulting in p.S95del is enclosed within a square.
Figure 3.
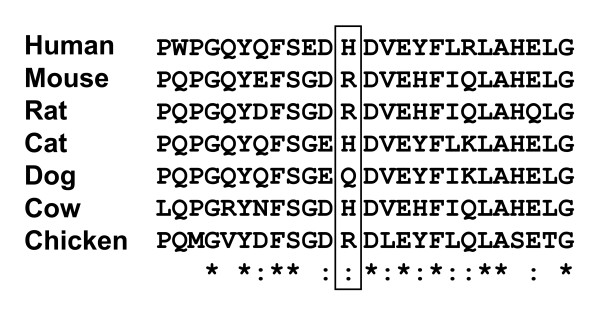
Alignment of the GLB1 homologs of six species around the H102 D variants indicated by squares http://www.ebi.ac.uk/Tools/clustalw/index.html. *Total sequence homology;: Very high homology;. High homology.
Transient expression of GLB1 plasmids in COS-1 cells
Expression patterns of these variant constructs in COS-1 cells were compared to those transfected with the normal GLB1 clone, pGS3 (Table 3). The GLB1 enzyme activities of cells transfected with pGS3-H102 D, pGS3-c.495_497delTCT, pGS3-A301V, and pGS3-G494V were 12%, 0%, 0%, and 0%, compared to those transfected with the control plasmid (pGS3), respectively.
Table 3.
β-Galactosidase activity measured in transiently transfected COS-1 cell lysates
| Vector name | Specific Activity (nmol/hr/mg of protein)1 | Percentage of wild-type activity2 |
|---|---|---|
| Controls | ||
| None | 320 ± 21.1 | 0 |
| pGS3 wt | 534 ± 27.7 | 100 |
| Variants | ||
| pGS3-H102D | 346 ± 19.4 | 12 |
| pGS3-c.495_497delTCT | 237 ± 14.5 | 0 |
| pGS3-A301V | 260 ± 20.3 | 0 |
| pGS3-G494V | 294 ± 27.5 | 0 |
1Enzyme activities are presented as average values of two independent experiments, as described in the Patients and Methods section.
2Specific activities were obtained after subtraction of activity in mock-transfected COS-1 cells measured as a baseline control in each experiment.
Discussion
More than 130 mutations in the GLB1 gene have been reported in GM1 or MBD cases from Japan, Northern/Southern America, and Europe. The majority of mutations appear to cluster in exons 2, 6, and 15 of the GLB1 gene [2]. These "hotspot" regions play important catalytic and/or regulatory roles. Here, we perform mutational analysis of two Han Chinese patients with GM1. Clinical manifestations of the patients can be distinguished according to the presence or absence of cardiac involvement. This atypical clinical feature has been reported in limited Caucasian patients. Three mutations in the GLB1 gene identified from the entire pathogenic allele of these two patients have not been reported previously. The variable severity of these clinical manifestations correlates with the mutation combinations. Patient 1, diagnosed with juvenile GM1 gangliosidosis without cardiomyopathy, is compound heterozygous for p.H102 D in exon 3 and p.A301V in exon 8, respectively. Although substitution of valine at position 301 with alanine seems to be homologous and non-deleterious, COS-1 cells expressing cDNA encoding p.A301V in vitro displayed no GLB1 enzyme activity. Low enzyme activity (~12% of the physiological level) was detected in COS-1 cells transfected with cDNA encoding the p.H102 D substitution. The p.H102 D appears to be a mild mutation responsible for the less severe phenotype of our juvenile patient displaying 2% residual enzyme activity. The p.A301V has been found in our patient 1 and in another juvenile patient [28], both presenting juvenile GM1 with central nervous system involvement and skeletal affection as well as absence of cardiac dysfunction and cherry red spots. The p.H102 D alone or combined with p.A301V can be related to hepatosplenomegaly.
Both c.495_497delTCT and p.G494V mutations identified in patient 2 led to absence of GLB1 enzyme activity in transient expression studies, correlating with lack of in vivo residual enzyme activity (< 1%). Two transcripts are processed from the primary β-galactosidase transcript, specifically, the classical lysosomal form of 677 amino acid β-galactosidase (GLB1) and the elastin binding protein (EBP) containing 546 residues in which exons 3, 4, and 6 are spliced out from pre-mRNA and exon 5 is frameshifted [5]. The frameshift in exon 5 results in a unique form of EBP, a 32-residue sequence containing an elastin/laminin-binding domain [15]. The two heterozygous mutations identified in patient 2 displaying cardiac involvement are localized in the GLB1 gene region common to the two transcripts, and thus affects both the lysosomal GLB1 enzyme and EBP protein. Both mutations can be related to infantile GM1 with cardiac dysfunction, as well as central nervous system involvement, cherry red spots and lack of skeletal affection.
The c.495_497delTCT mutation of the GLB1 transcript corresponds to a c.283_285delTCT variation of the EBP transcript, leading to in-frame deletion of an amino acid, p.Ser95del, in the elastin/laminin-binding domain of EBP. This variation in the elastin-binding domain of the EBP protein may directly impair tropoelastin binding to EBP and assembly of elastic fibers, which are consequently linked to the patient's cardiac involvement. Our data additionally support an association of EBP with cardiac involvement in GM1 gangliosidosis [16-19].
Conclusions
We identified three novel genetic mutations in the GLB1 coding region from Chinese patients with GM1. Patient 1 presented with the juvenile form of GM1 and 2% residual β-galactosidase activity in cultured fibroblasts. Expression of the two GLB1 mutants identified in patient 1, p.H102 D and p.A301V, in COS-1 cells led to 12% and 0% of normal activity. Patient 2 presented with the infantile form of GM1 and less than 1% of residual β-galactosidase activity in cultured fibroblasts. The two mutants, p.G949V and c.934-7del (p.L166del), identified from patient 2 were devoid of activity upon expression in COS-1 cells. The two mutations in patient 2 were localized in the GLB1 gene region common to lysosomal GLB1 and EBP, and could therefore affect both proteins. All the mutations correlated with the clinical manifestations of patients and residual β-galactosidase activity in cultured fibroblasts. While elastic fiber deposition requires further evaluation in patients, it is plausible that impaired elastogenesis and cardiomyopathy in GM1 gangliosidosis are caused by defects in the elastin-binding domain of EBP.
Abbreviations
GM1: GM1 gangliosidosis; GLB1: beta-galactosidases; EBP: elastin binding protein; PPCA: protective protein/cathepsin A; NEU1: neuraminidase.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JYW conceived and designed the study. CFY performed molecular analysis and transient expression, and drafted the manuscript. FJT participated in the design of the study and critically reviewed the draft. All authors have read and approved the final manuscript.
Contributor Information
Chi-Fan Yang, Email: cfyang@ibms.sinica.edu.tw.
Jer-Yuarn Wu, Email: jywu@ibms.sinica.edu.tw.
Fuu-Jen Tsai, Email: d0704@mail.cmuh.org.tw.
Acknowledgements
The authors are grateful to the patients and their parents for their participation in the study. This work was supported by China Medical University Hospital (grant number: DMR-92-079), Taichung, Taiwan and the National Science Council (grant number: NSC 89-2314-B-039-003), Taipei, Taiwan.
References
- Suzuki Y, Oshima A, Namba E. GM1-gangliosidosis and Morquio B disease. 8. New York: McGraw-Hill; 2001. [Google Scholar]
- Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab. 2008;17:391–396. doi: 10.1016/j.ymgme.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Hake CA, Martin BM, Kretz KA, Ahern-Rindell AJ, Naylor SL, Mudd M, O'Brien JS. Isolation, characterization, and mapping of a human acid beta-galactosidase cDNA. DNA Cell Biol. 1990;17:119–127. doi: 10.1089/dna.1990.9.119. [DOI] [PubMed] [Google Scholar]
- Morreau H, Bonten E, Zhou XY, D'Azzo A. Organization of the gene encoding human lysosomal beta-galactosidase. DNA Cell Biol. 1991;17:495–504. doi: 10.1089/dna.1991.10.495. [DOI] [PubMed] [Google Scholar]
- Morreau H, Galjart NJ, Gillemans N, Willemsen R, van der Horst GT, d'Azzo A. Alternative splicing of beta-galactosidase mRNA generates the classic lysosomal enzyme and a beta-galactosidase-related protein. J Biol Chem. 1989;17:20655–20663. [PubMed] [Google Scholar]
- Hinek A. Biological roles of the non-integrin elastin/laminin receptor. Biol Chem. 1996;17:471–480. [PubMed] [Google Scholar]
- Privitera S, Prody CA, Callahan JW, Hinek A. The 67-kDa enzymatically inactive alternatively spliced variant of beta-galactosidase is identical to the elastin/laminin-binding protein. J Biol Chem. 1998;17:6319–6326. doi: 10.1074/jbc.273.11.6319. [DOI] [PubMed] [Google Scholar]
- D'Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci USA. 1982;17:4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanba E, Tsuji A, Omura K, Suzuki Y. GM1-gangliosidosis: abnormalities in biosynthesis and early processing of beta-galactosidase in fibroblasts. Biochem Biophys Res Commun. 1988;17:794–800. doi: 10.1016/S0006-291X(88)80108-6. [DOI] [PubMed] [Google Scholar]
- van der Spoel A, Bonten E, d'Azzo A. Processing of lysosomal beta-galactosidase. The C-terminal precursor fragment is an essential domain of the mature enzyme. J Biol Chem. 2000;17:10035–10040. doi: 10.1074/jbc.275.14.10035. [DOI] [PubMed] [Google Scholar]
- Morreau H, Galjart NJ, Willemsen R, Gillemans N, Zhou XY, d'Azzo A. Human lysosomal protective protein. Glycosylation, intracellular transport, and association with beta-galactosidase in the endoplasmic reticulum. J Biol Chem. 1992;17:17949–17956. [PubMed] [Google Scholar]
- Bonten E, van der Spoel A, Fornerod M, Grosveld G, d'Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;17:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- Achyuthan KE, Achyuthan AM. Comparative enzymology, biochemistry and pathophysiology of human exo-alpha-sialidases (neuraminidases) Comp Biochem Physiol B Biochem Mol Biol. 2001;17:29–64. doi: 10.1016/S1096-4959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, Pshezhetsky AV. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat. 2003;17:343–352. doi: 10.1002/humu.10268. [DOI] [PubMed] [Google Scholar]
- Hinek A, Rabinovitch M, Keeley F, Okamura-Oho Y, Callahan J. The 67-kD elastin/laminin-binding protein is related to an enzymatically inactive, alternatively spliced form of beta-galactosidase. J Clin Invest. 1993;17:1198–1205. doi: 10.1172/JCI116280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrone A, Bardelli T, Donati MA, Giorgi M, Di Rocco M, Gatti R, Parini R, Ricci R, Taddeucci G, D'Azzo A, Zammarchi E. beta-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Hum Mutat. 2000;17:354–366. doi: 10.1002/(SICI)1098-1004(200004)15:4<354::AID-HUMU8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Hinek A, Wilson SE. Impaired elastogenesis in Hurler disease: dermatan sulfate accumulation linked to deficiency in elastin-binding protein and elastic fiber assembly. Am J Pathol. 2000;17:925–938. doi: 10.1016/S0002-9440(10)64961-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caciotti A, Donati MA, Boneh A, d'Azzo A, Federico A, Parini R, Antuzzi D, Bardelli T, Nosi D, Kimonis V. et al. Role of beta-galactosidase and elastin binding protein in lysosomal and nonlysosomal complexes of patients with GM1-gangliosidosis. Hum Mutat. 2005;17:285–292. doi: 10.1002/humu.20147. [DOI] [PubMed] [Google Scholar]
- Caciotti A, Donati MA, Bardelli T, d'Azzo A, Massai G, Luciani L, Zammarchi E, Morrone A. Primary and secondary elastin-binding protein defect leads to impaired elastogenesis in fibroblasts from GM1-gangliosidosis patients. Am J Pathol. 2005;17:1689–1698. doi: 10.1016/S0002-9440(10)61251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinek A, Rabinovitch M. 67-kD elastin-binding protein is a protective "companion" of extracellular insoluble elastin and intracellular tropoelastin. J Cell Biol. 1994;17:563–574. doi: 10.1083/jcb.126.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinek A. Nature and the multiple functions of the 67-kD elastin-/laminin binding protein. Cell Adhes Commun. 1994;17:185–193. doi: 10.3109/15419069409004436. [DOI] [PubMed] [Google Scholar]
- Hinek A, Wrenn DS, Mecham RP, Barondes SH. The elastin receptor: a galactoside-binding protein. Science. 1988;17:1539–1541. doi: 10.1126/science.2832941. [DOI] [PubMed] [Google Scholar]
- Hinek A, Keeley FW, Callahan J. Recycling of the 67-kDa elastin binding protein in arterial myocytes is imperative for secretion of tropoelastin. Exp Cell Res. 1995;17:312–324. doi: 10.1006/excr.1995.1321. [DOI] [PubMed] [Google Scholar]
- Pshezhetsky AV, Ashmarina M. Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog Nucleic Acid Res Mol Biol. 2001;17:81–114. doi: 10.1016/s0079-6603(01)69045-7. full_text. [DOI] [PubMed] [Google Scholar]
- Hinek A, Pshezhetsky AV, von Itzstein M, Starcher B. Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J Biol Chem. 2006;17:3698–3710. doi: 10.1074/jbc.M508736200. [DOI] [PubMed] [Google Scholar]
- Duca L, Blanchevoye C, Cantarelli B, Ghoneim C, Dedieu S, Delacoux F, Hornebeck W, Hinek A, Martiny L, Debelle L. The elastin receptor complex transduces signals through the catalytic activity of its Neu-1 subunit. J Biol Chem. 2007;17:12484–12491. doi: 10.1074/jbc.M609505200. [DOI] [PubMed] [Google Scholar]
- Hofer D, Paul K, Fantur K, Beck M, Burger F, Caillaud C, Fumic K, Ledvinova J, Lugowska A, Michelakakis H. et al. GM1 gangliosidosis and Morquio B disease: expression analysis of missense mutations affecting the catalytic site of acid beta-galactosidase. Hum Mutat. 2009;17:1214–1221. doi: 10.1002/humu.21031. [DOI] [PubMed] [Google Scholar]
- Hofer D, Paul K, Fantur K, Beck M, Roubergue A, Vellodi A, Poorthuis B, Michelakakis H, Plecko B, Paschke E. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clin Genet. 2010;17(3):236–246. doi: 10.1111/j.1399-0004.2010.01379.x. [DOI] [PubMed] [Google Scholar]
- Lin HC, Tsai FJ, Shen WC, Tsai CH, Peng CT. Infantile form GM1 gangliosidosis with dilated cardiomyopathy: a case report. Acta Paediatr. 2000;17:880–883. doi: 10.1080/080352500750043828. [DOI] [PubMed] [Google Scholar]
- Pan WH, Fann CS, Wu JY, Hung YT, Ho MS, Tai TH, Chen YJ, Liao CJ, Yang ML, Cheng AT, Chen YT. Han Chinese cell and genome bank in Taiwan: purpose, design and ethical considerations. Hum Hered. 2006;17:27–30. doi: 10.1159/000091834. [DOI] [PubMed] [Google Scholar]
- Ansaldi M, Lepelletier M, Mejean V. Site-specific mutagenesis by using an accurate recombinant polymerase chain reaction method. Anal Biochem. 1996;17:110–111. doi: 10.1006/abio.1996.0060. [DOI] [PubMed] [Google Scholar]
- Zhang S, Bagshaw R, Hilson W, Oho Y, Hinek A, Clarke JT, Callahan JW. Characterization of beta-galactosidase mutations Asp332-- > Asn and Arg148-- > Ser, and a polymorphism, Ser532-- > Gly, in a case of GM1 gangliosidosis. Biochem J. 2000;17(Pt 3):621–632. doi: 10.1042/0264-6021:3480621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbes M, D'Agrosa RM, Callahan JW. Human placental beta-galactosidase. Characterization of the dimer and complex forms of the enzyme. Biochem J. 1992;17(Pt 3):827–831. doi: 10.1042/bj2850827. [DOI] [PMC free article] [PubMed] [Google Scholar]