

Abstract
Chromatin structure is epigenetically altered via covalent modifications of histones to allow for heritable gene regulation without altering the nucleotide sequence. Multiple lines of evidence from rodents have established a role for epigenetic remodeling in regulating gene transcription in response to an altered gestational milieu. However, to date, it is unknown whether variations in the intrauterine environment in primates similarly induce changes in key determinants of hepatic chromatin structure. We hypothesized that a maternal high-fat diet would alter the epigenomic profile of the developing offspring, which would result in alterations in fetal gene expression. Age- and weight-matched adult female Japanese macaques were placed on control (13% fat) or high-fat (35% fat) breeder diets and mated annually over a 4-year interval. Fetuses in successive years were delivered near term (e130 of 167 days) and underwent necropsy with tissue harvest. Fetal histones were acid extracted for characterization of H3 modification and chromatin immunoprecipitation (ChIP) with differential display PCR; fetal RNA, DNA, and cytoplasmic and nuclear protein extracts were similarly extracted for comparison. Chronic consumption of a maternal high-fat diet results in a threefold increase in fetal liver triglycerides and histologic correlates of non-alcoholic fatty liver disease. These gross changes in the fetal liver are accompanied by a statistically significant hyperacetylation of fetal hepatic tissue at H3K14 (199·85±9·64 vs 88·8±45·4; P=0·038) with a trend towards the increased acetylation at H3K9 (140·9±38·7 vs 46·6±6·53; P=0·097) and at H3K18 (69·0±3·54 vs 58·0±4·04; P=0·096). However, epigenetic modifications on fetal hepatic H3 associated with gene repression were absent or subtle (P>0·05). Subsequent characterization of key epigenetic determinants associated with H3 acetylation marks revealed similar significant alterations in association with a high-fat maternal diet (e.g., relative fetal histone deacetylase 1 (HDAC1) gene expression 0·61±0·25; P=0·011). Consistent with our mRNA expression profile, fetal nuclear extracts from offspring of high-fat diet animals were observed to be significantly relatively deplete of HDAC1 protein(36·07±6·73 vs 83·18±7·51; P=0·006) and in vitro HDAC functional activity (0·252±0·03 vs 0·698±0·02; P<0·001). We employ these observations in ChIP differential display PCR to attempt to identify potential fetal genes whose expression is reprogramed under conditions of a high-fat maternal diet. We quantitatively confirm a minimum of a 40% alteration in the expression of several genes of interest: glutamic pyruvate transaminase (alanine aminotransferase) 2 (GPT2) (1·59±0·23-fold; P=0·08), DNAJA2 (1·36±0·21; P=0·09), and Rdh12 (1·88±0·15; P=0·01) are appreciably increased in fetal hepatic tissue from maternal caloric-dense diet animals when compared with control while Npas2, a peripheral circadian regulator, was significantly downmodulated in the offspring of high-fat diet animals (0·66±0·08; P=0·03). In this study, we show that a current significant in utero exposure (caloric-dense high-fat maternal diet) induces site-specific alterations in fetal hepatic H3 acetylation. Employing ChIP, we extend these observations to link modifications of H3 acetylation with alterations in gene-specific expression. These results suggest that a caloric-dense maternal diet leading to obesity epigenetically alters fetal chromatin structure in primates via covalent modifications of histones and hence lends a molecular basis to the fetal origins of adult disease hypothesis.
Introduction
According to Barker’s fetal origins of adult disease hypothesis, perturbations in the gestational milieu influence the development of diseases later in life (Barker 1995, Stein et al. 1996). This supposition gains support from animal models of nutritional constraint (Burdge et al. 2007, Gluckman et al. 2007, Lillycrop et al. 2007) and uteroplacental insufficiency-induced intrauterine growth restriction (IUGR; Fu et al. 2004, 2006, MacLennan et al. 2004, Ke et al. 2005) as well as epidemiologic studies (Klebanoff et al. 1989, Eriksson et al. 2001, Lesage et al. 2001, Bhargava et al. 2004, Rich-Edwards et al. 2005). These outcomes have been suggested to occur through the static reprograming of gene expression via alterations in the chromatin structure (epigenetic regulation). We and other researchers have previously demonstrated that uteroplacental insufficiency and IUGR in the rat results in covalent modifications of chromatin structure (Fu et al. 2004, 2006, MacLennan et al. 2004, Ke et al. 2005) or DNA methylation patterns (Burdge et al. 2007, Gluckman et al. 2007, Lillycrop et al. 2007) while leading to persistent changes in postnatal gene expression.
Genomic DNA exists endogenously in the nuclei of all eukaryotic cells as a highly folded, tightly compacted dynamic chromatin polymer wrapped around an octamer core of histones with a protruding charged 15–38 amino acid N terminus (termed a histone ‘tail’). These protruding tails influence nucleosome assembly into higher order chromatin structure. In its condensed state, chromatin folds so that the nucleosomes are stacked on one another, a structural configuration not readily accessible to gene activation (Jenuwein & Allis 2001, Li 2002, Bannister & Kouzarides 2005, Yan & Boyd 2006, Guenther et al. 2007). However, covalent modifications (i.e., acetylation, methylation, phosphorylation, poly-ADP ribosylation, and ubiquitination) of the H3 and H4 histone tails alter the interaction between histones and DNA, which affects nucleosome-to-nucleosome interactions as well as higher order chromatin folding. Hence, these post-translational modifications are often referred to as the so-called ‘histone code’ and likely enable the regulation of contact with the underlying DNA (Jenuwein & Allis 2001, Li 2002, Bannister & Kouzarides 2005, Morillon et al. 2005, Yan & Boyd 2006, Guenther et al. 2007).
Lysine methylation and acetylation of histones, particularly H3, targets and directs the epigenetic transcriptional machinery (Kasten et al. 2004, Pokholok et al. 2005). As such, it may be considered an important epigenetic ‘indexing system’ demarcating transcriptional active and inactive chromatin domains in the eukaryotic genome. While a hierarchical paradigm has yet to be established in primates, collective evidence from other eukaryotes suggests that while di/trimethylation of H3-K4 increases transcription, di/trimethylation of H3-K9 and trimethylation of H3-K27 repress transcription. This likely occurs by alternately facilitating or preventing binding of the epigenetic machinery via unique recognition sequences (chromodomains) with high specificity for methylation modifications on lysine residues (Li 2002, Morillon et al. 2005, Yan & Boyd 2006). Similarly, alternate recognition sequences (bromodomains) of effecter molecules associated with transcriptional activation have high affinity for H3 acetylation motifs at H3-K9 and H3-K14 (Kasten et al. 2004, Pokholok et al. 2005, Shogren-Knaak et al. 2006, Guenther et al. 2007). We and other researchers have previously demonstrated that such site-specific modifications and altered expression of key components of the epigenetic machinery occur in response to limited perinatal perturbations and are associated with persistent alterations in gene expression (Fu et al. 2004, 2006, MacLennan et al. 2004, Ke et al. 2005).
Despite our expanding understanding of the role of epigenetic modifications as they relate to models of IUGR, a paucity of information exists regarding these events in relation to maternal obesity. The current global health epidemic of obesity is considered a major contributor to the increasing prevalence of type II diabetes, as well as atherosclerotic, cardiovascular, and hypertensive morbidity and mortality (Must et al. 1999, Kopelman 2000, Field et al. 2001, Popkin & Gordon-Larsen 2004, Finkelstein et al. 2005, Rennie & Jebb 2005). In association with this rise in adult obesity throughout the developed and developing world, there occurs a disproportionate earlier onset of obesity among children (infants to adolescents; Strauss & Pollack 2001, Slyper 2004, Speiser et al. 2005). Given such anticipation (e.g., tendency of disease to appear at an earlier age of onset and with increasing severity in successive generations), the increased prevalence of childhood obesity cannot be attributed to either environment or genetics alone. Rather, the collective data from models of nutritional constraint and uteroplacental insufficiency would suggest that the gestational milieu profoundly influences the postnatal phenotype to render an increased susceptibility to childhood obesity via in utero alterations in fetal histone [H3] covalent modifications (Klebanoff et al. 1989, Eriksson et al. 2001, Lesage et al. 2001, Bhargava et al. 2004, Fu et al. 2004, 2006, MacLennan et al. 2004, Ke et al. 2005, Rich-Edwards et al. 2005).
We therefore hypothesized that a maternal high-fat diet would alter fetal hepatic chromatin structure, as well as the functional expression of key components of the epigenetic machinery. Following identification of changes in site-specific histone H3 acetylation signatures associated with transcriptional activation in other model systems, we further hypothesized that the employment of chromatin immunoprecipitation (ChIP) would lead us to those fetal genes whose expression is altered, or ‘reprogramed’, under conditions of maternal obesity. With confirmation of the persistent differential expression of such genes postnatally, we speculate that these observations will lead to the identification of patterns of modifiable gene expression integral to the maintenance of glucose, lipid, and fatty acid homeostasis, which sets the stage for obesity.
Materials and methods
Animals
All animal procedures were in accordance with the guidelines of Institutional Animal Care and Use Committee of the Oregon National Primate Research Center, the University of Utah, and Baylor College of Medicine. Briefly and as described in further detail elsewhere (Bishop et al. 2008), age- and weight-matched young (5–6 years) adult female Japanese macaques were socially housed in indoor/outdoor enclosure in groups of five to nine with one to two males per group. Animals were separated into two groups: control-fed standard monkey chow that provides 13% of calories from fat (Monkey Diet no. 5052, Lab Diet, Richmond, IN, USA) and the high-fat diet group providing 35% of calories from fat (Custom Diet 5A1F, Test Diet, Richmond, IN, USA) and included calorically dense treats. Prior to this study, all animals were naïve to any experimental protocols. Animals were allowed to breed naturally over four successive seasons to yield offspring of ‘years 1’, ‘2’, ‘3’, or ‘4’. Pregnancies terminated by Cesarean at gestational day 130 (e130 as confirmed by ultrasound with biometry). Immediately after cesarean delivery, the fetuses were delivered to necropsy, deeply anesthetized (sodium pentobarbital >30 mg/kg i.v.) and then exsanguinated. Harvested tissue was dissected and flash-frozen in liquid nitrogen or fixed in 4% paraformaldehyde in phosphate buffer (pH 7·4). In such a manner, all animal tissue used in these studies was graciously supplied in collaboration with Dr Kevin Grove and the Oregon National Primate Research Centre (ONPRC) (Bishop et al. 2008). All studies reported herein employed snap-frozen fetal liver from control diet (n=9) or high-fat diet (n=10)-fed dams for 1 or 2 years by the above-described criteria. For the purpose of Japanese macaque species-specific cDNA cloning, tissue from corralled (non-experimental protocol) animals were employed.
Histone isolation and western blotting
Histones were isolated from day 0 fetal liver tissue by acid extraction as previously described (Fu et al. 2004, 2006, Ke et al. 2005). Histone concentrations were determined using the micro BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Histones (20 μg) were separated on 15% SDS-PAGE gels and transferred by electroblotting to polyvinyldifluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Blocking was carried out with freshly prepared TBS–Tween plus 3% non-fat milk (TBST–MLK). After washing, the membrane was incubated overnight with primary antibodies diluted in TBST–MLK. Primary antibodies were as follows: anti-AcH3K9 1:400 (Cell Signaling, Beverly, MA, USA), anti-AcH3K14 1:5000, anti-AcH3K18 1:500, and anti-histone H3 1:2000 (Upstate Cell Signaling Solutions, Lake Placid, NY, USA). Appropriate secondary antibodies conjugated with horse-radish peroxidase (HRP) were incubated for 1 h at room temperature. Signals were detected using electrochemiluminescent (ECL) performed according to the manufacturer’s instructions (Amersham). The amount of acetylated H3 was quantified by normalization to the amount of total H3 in each sample; thus, relative arbitrary densitometry units are reported. Though most studies focusing upon histone covalent modifications do not perform this step of normalization to total H3, we elected to utilize such an internal loading control with in vivo tissues that are potentially more heterogeneous in terms of histone acetylation than cell culture or clonal tumor cell lines.
Acid urea gel
We employed a modification of the original method described by Pastewka et al. (1973) for haptoglobin separation. Briefly, the stacking gel protocol was modified to include dissolving 3·84 g urea in 1 ml formate buffer (pH 3·2; 0·48 g NaOH, 1·56 ml of 88% formic acid, QS to 100 ml), 2 ml of 10% stacking gel reagent (10% acrylamide, 2·5% bisacrylamide), and ddH2O to 6 ml; 26 μl of 10% ammonium persulfate and 20 μl tetramethylethylenediamine (TEMED) were added. The resolving gel preparation was done as previously described with the addition of 200 μl of 10% ammonium persulfate and 30 μl TEMED to a total volume of 35 ml (Pastewka et al. (1973)). The gel was pre-run for 4–6 h at 200 Vunder reversed lead conditions in electrode buffer (pH 3·7; 0·4 M glycine–0·05 M formic acid). Acid-extracted histones (50 μg) were diluted in 9·6 M urea with 2 μl basic fuschin tracking dye and loaded. The gels were run at 18 mA overnight at 4 °C.
For western blotting, gels were washed twice for 30 min in Wash Buffer I (0·5% w/v SDS and 50 mM acetic acid), then for 30 min in Wash Buffer II (50% 2-ME, 2% w/v SDS, 62·5 mM Tris–HCl, pH 6·8). Transfer onto PVDF occurred in Transfer Buffer (20% v/v methanol, 25 mM CAPS–NaOH, pH 10·0) at 35 V overnight at 4 °C. The remainder of the blotting and quantification is as described above for the detection of histone modifications by western blotting.
RNA isolation
Total RNA was extracted from fetal hepatic tissue using the RNeasy Mini Kit (Qiagen), treated with DNase I (Qiagen), and then quantified. Gel electrophoresis confirmed the integrity of the samples.
cDNA cloning from Japanese macaque
RNA was extracted from fetal hepatic tissue of corral (non-experimental protocol) animals. cDNA was synthesized from 5 μg DNase-treated total RNA employing the SuperScriptIII First-Strand Synthesis system for RT-PCR by the manufacturer’s directions (Invitrogen). Primers and probes employed in cloning were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH): for (5′-ATGGGGAAGGTGAAGGTYGG-3′), rev (5′-GGTCGTTGAGGGCAATGC-3′); DNA methyltransferase 1 (Dnmt1): for (5′-TCAGACTGGCGCGATCTG-3′), rev (5′-CTGCTGAGGCACTCTCTCG-3′); histone deacetylase 1 (HDAC1): for (5′-GACGGACATCGCTGTGAATTG-3′), rev (5′-TCACTTGATCTTCTCCAGGTACTC-3′); Npas2: for (5′-AAGTCTGAGAAGAAGCGTCGG-3′), rev (5′-GGCAGGTATCCTAT-3′); DNAJA2: for (5′-GACACGAAGCTGTACGACATC-3′), rev (5′-CGAGTGCTATCAAATTCCTGAAGC-3′); GPT2: for (5′-GAGTCCATGAACCCGCAGG-3′), rev (5′-GCGTCGAGCTCAGAGATGAC-3′); and Rdh12: for (5′-AGCTCCATCCATCAGGAAGTTC-3′), rev (5′-TCCTCTTGCAGTCACTGAAGTAC-3′). PCR products were cloned into plasmid vectors using the TA cloning kit (Invitrogen). Multiple colonies derived from the cloning procedure were sequenced according to the manufacture’s instructions for double-stranded plasmid DNA using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) at the UUHSC Core Sequencing Facility.
Real-time RT-PCR
cDNA was synthesized using the same method as previously described. Briefly, mRNA levels of respective genes in independent samples were quantitated using the (ΔΔCT) method of quantitation as described by others (Pfaffl 2001). GAPDH was used as internal control. The primers and probes sequence are shown: methyl CpG binding protein 2 (MeCP2): for (5′-GTTAGGGCTCAGGGAAGAAAAGTC-3′), rev (5′-TTCTTCACCTTTTTAAACTTGAGGG-3′), probe (5′-CAGGATCTCCAGGGCCTCAAGGACA-3′); HDAC1: for (5′-TGACGAGTCCTATGAGGCCAT-3′), rev (5′-CCGCGCTAGGTTGGAACAT-3′), probe (5′-CCATTACTTTGGACATGACCGGCTTGA-3′); Dnmt1: for (5′-ACACCTACCGGCTCTTCGG-3′), rev (5′-TTGATCTCCAAGCCAATGGC-3′), probe (5′-AACATCCTGGACAAGCACCGGCAG-3′); GAPDH: for (5′-GGGAAGGTGAAGG TCGGAGT-3′), rev (5′-AAGCAGCCCTGGTGACCA-3′), probe (5′-ACGGATTTGGTCGTATTGGGCGC-3′); GPT2: for (5′-GGCAGCAGCCAATCACCTT-3′), rev (5′-GTCCAGCAGGTTTGGGTAGG-3′), probe (5′-CTCCGGCAGGTAATGGCACTGTGC-3′); Rdh12: for (5′-TGGCAAGGAGACGGCTAGA-3′), rev (5′-CTTCAGTACATCTCTGCAGGCAA-3′), probe (5′-TTGCTAGCCGAGGAGCCCGAGTCT-3′); DNAJA2: for (5′-CCCTGGCAAAGTAATTGAACCA-3′), rev (5′-AAGGGATTACGATACTGCGGC-3′), probe (5′-TCCCTTCACCTCGAACTACACGAACACCT-3′); and Npas2: for (5′-CGGCAGCATCATCTATGTCTCT-3′), rev (5′-GATCCATGACATCCGACGGT-3′), probe (5′-ACAGTATCACGCCTCTCCTTGGGCATT-3′).
Protein isolation and western blotting
Flash-frozen hepatic tissue samples (1 cm3) were ground under liquid nitrogen. Ground tissue was thereafter rinsed in ice-cold PBS to lyse any red blood cells. For the preparation of nuclear extracts, tissue was resuspended in 5 volumes of ice-cold lysis Buffer C (1 mM dithiothreitol (DTT), 0·5 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM KCl, 10 mM HEPES, pH 7·9, 1·5 mM MgCl2) and incubated for 30 min. After centrifugation at 2000 g at 4 °C for 5 min, the cell pellet was again resuspended in 2 vol of Buffer C with 20 strokes of a Dounce homogenizer. The lysate was centrifuged at 1000 g for 10 min. The nuclear pellet was resuspended in 0·5 ml Buffer D (1·5 mM MgCl2, 1 mM DTT, and 420 mM NaCl, 5 mM PMSF, 25% (v/v) glycerol, 0·2 mM EDTA, 20 mM HEPES, pH 7·9), incubated at 4 °C for 30 min, and then centrifuged at 17 000 g for 15 min. The collected supernatants were stored at −80 °C until use. For cytosolic extracts, 1 cm3 fetal hepatic samples were homogenized in ice-cold lysis buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7·4, 1 mM EGTA, 0·25% Na-deoxycholate, and 1% Igepal CA-630) with protease inhibitors and 0·1 M PMSF. After centrifugation at 10 000 g at 4 °C for 15 min, supernatants were stored at −80 °C until use.
Protein concentrations were determined by the BCA method (Pierce). Proteins were separated by 10% SDS-PAGE ready gels (Bio-Rad) and transferred onto nitrocellulose membranes in standard transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol). After blocking the membranes with 5% milk in TBS for 1 h, bound proteins were exposed to specific antibodies against DNMT1, HDAC1, or MeCP2 (rabbit polyclonal, Santa Cruz Biotechnology) overnight at 4 °C. After extensive washing in TBS with 0·1% Tween-20 (TBST), a 1/2000 dilution of goat anti-rabbit HRP secondary antibody (Cell Signaling Technology, Inc.) was applied and incubated for 1 h at room temperature. After extensive washing in TBST, signals were detected with Western Lightning ECL (Perkin–Elmer Life Sciences, Inc., Waltham, Massachusetts, USA) and Biomax film (Amersham) and quantified by densitometry or by quantification on a Kodak Image Station 2000R (Eastman Kodak/SIS).
HDAC enzymatic activity
The Biomol HDAC Colorimetric Activity Assay (AK-501) was utilized under manufacturer’s suggestions with nuclear extracts under the above-described conditions (Biomol Research Laboratories, Inc.; Plymout Meeting, PA, USA).
ChIP assay
The protocol, as previously described, was used with the following modifications (Ke et al. 2005, Fu et al. 2006). One hundred milligrams of day 0 fetal hepatic tissue were ground in liquid nitrogen and fixed with formaldehyde to a final concentration of 1% for 10 min. The chromatin was sonicated (Fisher Scientific, Sonic Dismembrator, Model 100, Pittsburgh, PA, USA) 12 times for 10 s on ice at the highest level to generate chromatin fragments of 500–2000 bp. The sonicated chromatin was quantified on the basis of DNA content at A260. The chromatin equivalent of 40 μg DNA based on the absorption at A260 was used in each immunoprecipitation (IP) reaction. In order to determine the amount of antibody to use to immunoprecipitate formaldehyde cross-linked chromatin, each antibody was titrated against 24 μg acid-extracted histone protein and 48 μg sonicated chromatin in 110 μl IP dilution buffer. For anti-acetyl H3/K14 (Upstate Cell Signaling Solutions), an optimal previously determined antibody concentration of 20 μg was employed. The DNeasy Tissue Kit (Qiagen) was used to purify the DNA from the total amount of DNA extracted from either 40 μg chromatin or immunoprecipitated from 40 μg chromatin using anti-acetyl H3/K14, anti-rabbit IgG-HRP, and no antibody respectively. The latter two groups were run to verify the specificity of the ChIP antibodies. DNA was quantitated by measuring A260/A280.
Differential display PCR (DD-PCR)
A total of five 16 mer CG-rich arbitrary primers were used for differential display. ChIP DNAs (50 ng) from offspring of control and high-fat maternal diet were used as independent samples in the DD-PCR. The PCR used 1× PCR Gold Buffer (Applied Biosystems), 2·5 mM MgCl2, 2·5 μCi [α-33P]dCTP (Perkin–Elmer Life Sciences), 500 nM primers, and 1·25 units Ampli Taq Gold (Applied Biosystems). Initial denaturation was performed at 95 °C for 10 min, followed by 40 cycles of 94 °C for 30 s, 44 °C for 60 s, and 72 °C for 90 s for primers 315–317; and annealing temperature 50 °C for primers 318 and 319. PCR product (5 μl) was added to 1 μl loading dye (95% formamide, 20 mM EDTA, and 0·05% each bromophenol blue and xylene cyanol). After the samples were heated to 94 °C for 3 min, and then immediately cooled on ice, 4 μl of each sample was loaded onto a 0·4 mm thick, 34 cm long 5% polyacrylamide sequencing gel under denaturing conditions (7 M urea) and migrated in 1× Tris–borate–EDTA for about 5 h at 1500 V, 70–100 mA (to keep the temperature of the gel around 50 °C). After being dried, the gels were exposed to X-ray films at −80 °C.
DD-PCR primers used were as follows noted for primer sets ‘A’, ‘B’, and ‘C’: ‘A’: 5′-TGCCTCGAGARGCYCG-3′, 5′-TGCCTCGAGAYGCRCG-3′; ‘B’: 5′-TGCCTCGAGAGGCYCG-3′, 5′-TGCCTCGAGAGGYCCG-3′; ‘C’: 5′-AAGCTTGAGAGRCGRI-3′; 5′-TGCCTCGAGAYGCRC-3′.
Isolation and sequencing of DNA fragments
Candidate bands that were differentially represented between offspring of control and high-fat maternal diets were excised from dried polyacrylamide gels and placed in a microcentrifuge tube containing 50 μl sterile H2O. The microcentrifuge tube was then heated to 80 °C for 10 min and vortexed to facilitate dissolution of DNA. The eluate (2 μl) was then used in PCR with the same primers used in the original DD-PCR to generate sufficient amounts of template for plasmid cloning and sequencing. PCR ingredients and amplification parameters were the same as described above. PCR products were cloned into plasmid vectors using the TA cloning kit (Invitrogen). Multiple colonies derived from the cloning procedure were sequenced according to the manufacturer’s instructions for double-stranded plasmid DNA using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). The resulting nucleotide sequences were then compared with the GenBank sequences, using the BLAST multispecies alignment database (NCBI site http://www.ncbi.nih.gov/Sitemap/index.html).
Statistical analysis
All data represent the mean±S.E.M. Mean values were compared using Fisher’s independent samples two-tailed student’s t-test or a paired samples t-test for equality of means and determination of statistical significance.
Results
Fetal histone H3 undergoes modification in response to a maternal high-fat diet
DNA methylation and histone modifications serve as epigenetic marks for active or inactive chromatin in a variety of model systems. Other investigators profiling nucleosome modifications across yeast and mammalian genomes in an effort to produce high-resolution genome-wide maps of histone acetylation and methylation have identified H3 lysine 9 and 14 acetylation (AcH3K9 and AcH3K14) as key modifications correlating with active gene transcription (Jenuwein & Allis 2001, Li 2002, Bannister & Kouzarides 2005, Yan & Boyd 2006, Guenther et al. 2007). In higher eukaryotes, AcH3K9 and AcH3K18 occur in intergenic regions of expressed genes (Kurdiastani et al. 2004, Bernstein et al. 2005, Roh et al. 2005). Similarly, peaks of H3K4 di- and trimethylation (H3K4me2 and me3) correlate with gene transcription: H3K4me3 punctuates 5′ transcriptional start sites while H3K4me2 appears to reside elsewhere in the vicinity of active genes (Santos-Rosa et al. 2002, Kurdiastani et al. 2004, Pokholok et al. 2005, Roh et al. 2005, Guenther et al. 2007). By contrast, inactive genes have been shown to alternately lack these modifications and manifest distinct patterns of repressive methylation marks (H3K9me2 and me3, H3K27me2me3; Martens et al. 2005). While these studies by other investigators have utilized ChIP-on-chip methodologies for their characterizations, unavailability of macaque-specific arrays make similar high-throughput approaches in our model system impossible. For these reasons, we sought to characterize these key H3 modifications via western blot analyses in the primate fetal epigenome of offspring of adult animals exposed to a caloric-dense or control diet for 2 years (Bishop et al. 2008).
Western blotting with antibodies specific to H3AcK9, H3AcK14, and H3AcK18 was employed to determine the relative degree of lysine-specific acetylation after normalization to total histone H3 (Fig. 1a). As shown in Fig. 1a, exposure to a maternal high-fat diet resulted in a statistically significant hyperacetylation of fetal hepatic tissue at H3K14 (relative arbitrary densitometry units of 199·85±9·64 vs 88·8±45·4; P=0·038) with a trend towards the increased acetylation at H3K9 (relative arbitrary densitometry units of 140·9±38·7 vs 46·6± 6·53; P=0·097) and at H3K18 (relative arbitrary densitometry units of 69·0±3·54 vs 58·0±4·04; P=0·096). When we similarly examined epigenetic modifications on fetal hepatic H3 alternately associated with repression (dimethylation of H3K9 (H3K9me2) or trimethylation of H3K9 (H3K9me3) and ditrimethylation of H3K27 (H3K27me2me3); Barski et al. 2007) versus activation (di- and trimethylation of H3K4; Guenther et al. 2007), differences after normalization to total H3 were non-significant (Fig. 1b). Thus, fetal hepatic H3 methylation of H3K4me2 (relative arbitrary densitometry units of 119·0±7·43 vs 115·7±6·96; P=0·765), H3K9me2 (relative arbitrary densitometry units of 435·5±44·24 vs 323·3±27·21; P=0·107), H3K9me3 (relative arbitrary densitometry units of 278·7±14·16 vs 244·0±11·06; P=0·130), and H3K27me2me3 (relative arbitrary densitometry units of 446·5±52·89 vs 305·0±9·29; P=0·075) were minimally and non-significantly altered under conditions of a maternal high-fat diet when compared with control diet.
Figure 1.

Fetal histone H3 undergoes modification in response to a maternal high-fat diet. (a) Western blotting with antibodies specific to H3AcK9, H3AcK14, and H3AcK18 and (b) H3K4me2, K9me2, K9me3, and K27me2me3 were employed to determine the relative degree of lysine-specific acetylation (a) and methylation (b) after normalization to total histone H3. Histones were acid extracted from fetal hepatic tissue from control (n=3) and high-fat diet (n=4)-exposed animals in year 2, and resolved on SDS-PAGE gels. Relative densitometry was used in quantification (mean±S.E.M). Shown are representative bands of a minimum of three separate experiments. (c) Acid-extracted histones were resolved on an acidic urea gel under conditions of reverse polarity to enable resolution of post-translationally modified histone variants by virtue of their charge difference. Thus, retardation reflects increasing modified acetylation states following western blotting (e.g., anti-H3AcK14, left panel). Relative densitometry was used in quantification for the determination of the ratio of the arbitrarily designated ‘A’ versus ‘B’ variants AcK14A/B (mean± S.E.M). Shown are the representative bands of a minimum of three separate experiments.
Given our significant observed increased acetylation at H3K14, we next investigated the states of acetylation among histones in the fetal primate epigenome with resolution of modified variants of acid-extracted histones on an acidic urea gel (Pastewka et al. 1973). Briefly, acid-extracted histones were resolved on 10% urea gels (pH 4·6) to enable the separation of differentially modified histone variants by the virtue of charge and mass. With subsequent quantification employing modification-specific antibodies via standard western blot (i.e., detection with an anti-AcK14 antibody), we were able to differentiate whether observed histone modifications were indicative of radical changes in global acetylation as detected by western blot, or rather represented an ordered change in the ratio of modified (increasing) acetyl variants. As observed in Fig. 1c, employment of this technique indeed identified an altered ratio of modified acetyl variants, where the upper band (designated as ‘AcK14A’) is relatively greater in fetal hepatic tissue following in utero exposure to a maternal high-fat diet than the lower band (‘AcK14B’). By contrast, western blotting with the other antibodies employed in Fig. 1a and b failed to detect significant alterations in the migration pattern under identical conditions (data not shown). It is of further interest to note that while we cannot state what the variants AcK14A and AcK14B are per se, their migration patterns run in presumptive intermediate states of modified increasing acetylation, as defined by the virtue of their co-migration with the banding patterns of H3K4me2, yet subtly retarded migration from AcH4K16 (data not shown; Santos-Rosa et al. 2002, Shogren-Knaak et al. 2006). Thus, while an altered gestational milieu does not radically alter global fetal hepatic acetylation, modified H3 acetyl variants consistent with an increasing extent of acetylation are observed (Fig. 1c).
In the light of our collaborators’ observations of differential maternal sensitivity and resistance to the high-fat diet (Bishop et al. 2008), it is of interest to note that this is not an absolute finding and while modified acetylation occurs in the majority of offspring among the high-fat diet-exposed animals, there is individual variation (lanes 5–7 versus lane 4 versus lanes 1–3). Nevertheless, when a mean ratio of AcK14A/B is calculated, a significant difference of 5·15±1·03 vs 1·53±0·04 is observed under conditions of high-fat diet exposure (P=0·032).
A maternal high-fat diet modifies expression of fetal hepatic epigenetic determinants related to H3 acetylation
In mammals, DNA methylation is catalyzed by several (lysine) site-specific Dnmts (Dnmt1, Dnmt3a, and Dnmt3b) essential to embryonic development; Dnmt1 is generally considered a maintenance enzyme and has been linked with the regulation of multiple biologic processes (Jenuwein & Allis 2001). The methyl-CpG-binding protein 2 (MECP2) binds methylated DNA in vivo, and when complexed with HDAC has a high specificity for methylated CpG islands; deficiency of functional MeCP2 causes insufficient recruitment of the mSinA/HDAC1, HDAC2 complex to adequately repress transcription. HDAC1 is a class I HDAC, and in the process of silencing gene expression HDAC1-Dnmt1 complexes initiate the recruitment of methyl-CpG-binding domain proteins that mediate gene silencing by facilitating CpG island hyper-methylation. Of interest, recent evidence in mammalian systems demonstrate that these multimeric complexes show specificity for histone marks associated with gene induction (e.g., H3K14 acetylation; Li 2002, Morillon et al. 2005, Yan & Boyd 2006, Schnekenburger et al. 2007). Of interest, there is no published data to date in higher eukaryotes describing the relative role of MeCP2 or Dnmt1 overexpression in enabling for the transcriptionally permissive chromatin structure (PubMed, 1996–current).
Given our observed site-specific modifications in H3K14 acetylation, we sought to determine whether exposure to a high-fat diet in utero resulted in alterations in the expression or function of these aforementioned key epigenetic determinants. Given that the Japanese macaque genome has not yet been sequenced, we first cloned the cDNA for MeCP2, Dnmt1, and HDAC1 from Japanese macaque liver based on aligned murine and human sequences to identify conserved 5′ regions with multiple exons. Given a high degree of identity to the human genome (99, 95, and 96% respectively), real-time primers and probes at exon junction(s) were designed from human 5′ sequences bearing an exact match to cloned macaque sequence. As shown in Fig. 2a and consistent with our findings in Fig. 1 and those of other investigators (Lillycrop et al. 2007), we failed to observe a significant change in mean fetal hepatic mRNA expression over offspring of control diet animals for MeCP2 (1·68±0·35-fold; P=0·151). However, significant modest increases in expression of Dnmt1 (1·88±0·21 fold; P=0·05) and decreased expression of HDAC1 mRNA occurred in offspring of animals exposed to a high-fat diet (0·61±0·25; P=0·011). Moreover, when we similarly examined gene expression differences in a limited number of offspring allowed to go to term and then killed at postnatal day 30 (n=3) following in utero exposure to maternal high-fat or control diet for 2 years, we observed a persistent alteration in affected gene expression. Thus, HDAC1 remained downregulated in its expression (0·76±0·08; P=0·03) in high-fat diet-exposed offspring at postnatal day 30.
Figure 2.

A maternal caloric-dense diet modifies expression of fetal hepatic epigenetic determinants. Given our observed site-specific modification in H3 acetylation, we quantitated alterations in the expression and activity of key epigenetic determinants in the offspring of high-fat-fed animals. (a) Real-time RT-PCR estimate of relative expression of MeCP2, Dnmt1, and HDAC1 of high-fat (n=9) relative to control (n=10) maternal diet following normalization to GAPDH (ΔΔCT). Data are plotted as the mean ΔΔCT of high-fat diet-exposed animals in separate experiments (circles) relative to control diet offspring (dotted line); bars are thus the mean derived from a minimum of three separate experiments. (b) Quantification of fetal hepatic MeCP2, Dnmt1, and HDAC1 protein expression (left panel), and HDAC nuclear in vitro enzymatic activity (right panel). One hundred micrograms of cytosolic (Dnmt1) or nuclear (MeCP2 and HDAC1) protein were resolved on 2–12% SDS-PAGE gels, and quantification of protein was performed by western blotting. Relative densitometry following normalization to GAPDH was performed and protein expression values are plotted as mean±S.E.M. For the determination of HDAC enzymatic activity, 80 μg nuclear extracts were assayed for HDAC enzymatic activity using previously described methods. Data are plotted as mean±S.E.M. absorbance at 405 nm for control (n=3) and high-fat diet (n=4) offspring.
No significant alteration in expression of nuclear MeCP2 protein (36·04±12·54 vs 15·08±3·70; P=0·225) nor cytosolic Dnmt1 (31·97±4·08 vs 21·79±2·67; P=0·114) occurred in fetal hepatic extracts (Fig. 2b). Consistent with our mRNA expression profile, fetal nuclear extracts from offspring of high-fat diet animals were observed to be significantly relatively deplete of HDAC1 protein (36·07±6·73 vs 83·18±7·51; P=0·006) and in vitro HDAC functional activity (0·252±0·03 vs 0·698±0·02; P<0·001).
ChIP with differential display PCR
In an effort to identify genes that might undergo reprograming in response to histone modifications diagnostic for transcriptionally permissive chromatin, we employed ChIP with an antibody against AcH3K14. We were further aided in our decision to utilize this modification motif based on our results presented in Figs 1 and 2 demonstrating significant alterations in H3 acetylation and its determinants but not methylation per se.
Briefly, antibodies to AcH3K14 were used to precipitate 1% formaldehyde-stabilized fetal hepatic histone–DNA complexes from offspring of control and high-fat diet animals. DNA that was in complex with modified chromatin was subsequently amplified with a modified series of degenerate PCR primers and resolved on a urea gel; differences (either up or downregulated) among both individual offspring as well as diet-clustered groups were observed (d.n.s). Distinct bands appreciably variant by maternal diet were excised, and eluted DNA was cloned to enable BLAST against a multispecies genomic database. Of the 68 sequenced clones from the AcH3K14 ChIP, 32% were identified as genomic intergenic sequences, while 53% matched to 5′ regions of transcribed mammalian genes; 15% were not identifiable against the database (data not shown).
Based on the anticipated postnatal phenotype and blinded parallel microarray gene expression data (Bishop et al. 2008), four AcH3K14 ‘reprogramed’ genes were deemed to be of potential interest: GPT2 (an alanine aminotransferase, NCBI reference sequence NM_133443), DNAJA2 (an hsp70 co-chaperone, NCBI reference sequence NM_005880), Rdh12 (an all trans and 9-cis retinol dehydrogenase responsive to oxidative stress, NCBI reference sequence BC025724), and Npas2 (a hepatic forebrain paralog of Clock, NCBI reference sequence NM_002518). To verify that quantitative alterations in fetal hepatic mRNA expression occurred in response to a maternal high-fat diet perturbation, the Japanese macaque cDNA was cloned and real-time PCR primers and probes were designed accordingly. As shown in Fig. 3, we quantitatively confirm a minimum of a 40% alteration in the expression of each of these genes. Thus, GPT2 (1·59±0·23-fold; P=0·08) and DNAJA2 (1·36±0·21; P=0·09) trend towards significance, and Rdh12 (1·88±0·15; P=0·01) was appreciably significantly increased in fetal hepatic tissue from maternal caloric-dense diet animals when compared with control (Fig. 3). Of interest, Npas2 was significantly downmodulated in the offspring of high-fat diet animals (0·66±0·08; P=0·03). Consistent with this observation, its negative regulatory counterpart Cry1 is overexpressed under conditions of a maternal high-fat diet (1·75±0·28, P=0·03; data not shown). When we again examined gene expression differences in a limited number of offspring allowed to go to term and then killed at postnatal day 30 (n=3), following in utero exposure to maternal high-fat or control diet for 2 years we observed a persistent but non-significant trend in affected gene expression. Thus, Npas2 trended towards a downregulation in its expression (0·53±0·31; P=0·22) and Cry1 trended towards overexpression (1·45±0·61; P=0·14) in high-fat diet-exposed offspring at postnatal day 30, a finding that likely did not reach statistical significance due to small sample size.
Figure 3.
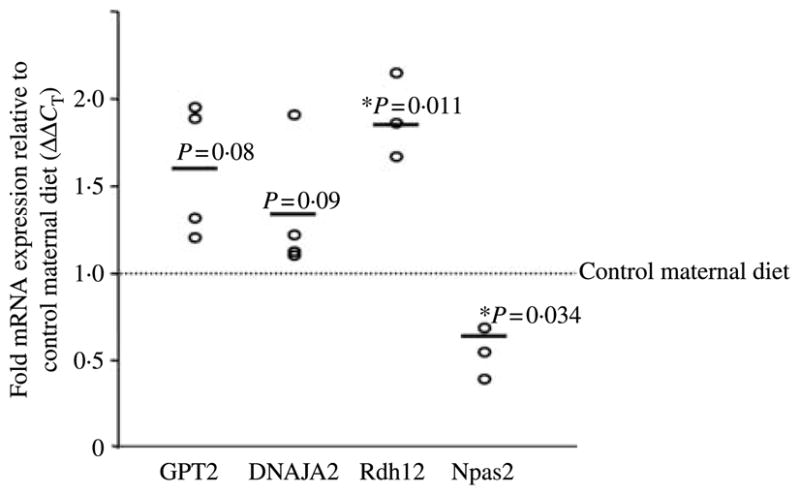
Quantification of fetal hepatic mRNA expression of ‘reprogramed’ primate genes under conditions of a maternal high-fat diet. Real-time RT-PCR primers and probes were designed from cloned Japanese macaque sequence for GPT2, DNAJA2, Rdh12, and Npas2. Quantification of fetal hepatic mRNA expression is shown as fold mRNA (mean±S.E.M.) expression among high-fat (n=9) relative to control maternal diet (n=10)-exposed offspring following normalization to GAPDH ΔΔCT). Data are plotted as the mean ΔΔCT of high-fat diet-exposed animals in separate experiments (circles) relative to control diet offspring (dotted line); bars are thus the mean derived from a minimum of three separate experiments.
Alterations in fetal hepatic heat shock protein expression under conditions of an in utero high-fat maternal diet
Parallel utilization of microarray analysis (Bishop et al. 2008) and ChIP with differential display (Fig. 3) revealed a number of anticipated changes in the expression or remodeling of genes involved in fatty acid catabolism and ketone body synthesis (e.g., GPT2), detoxification of lipid peroxidation products in response to oxidative stress (e.g., Rdh12), and the coordinate regulation of peripheral metabolism and satiety signals with circadian mechanisms (e.g., Npas2). Interestingly, the molecular chaperones (the so-called ‘heat shock proteins’, Hsps) and co-chaperones (e.g., DNAJA2) are postulated to be responsible for the repairs of misfolded or damaged proteins under oxidative and lipotoxic environments, and under both genomic screening approaches Hsps were observed to be appreciably altered in their expression in offspring of high-fat diet-fed animals (Kim et al. 2004, Ripperger & Schibler 2006). To further characterize the potential implications of alterations in fetal Hsp gene expression with the anticipated postnatal obese pathophysiology, we sought to quantify transcriptional and translational expressions of additional integral Hsp family members (Kim et al. 2004). As shown in Fig. 4a, mRNA expression of DNAJA2 transcriptionally regulated Hsp27 (2·17±0·25; P=0·003) and Hsp70 (1·99±0·28; P=0·025) was significantly increased in fetal hepatic tissue from maternal caloric-dense diet in years 1 and 2 (n=17) when compared with control diet (n=9). Similarly, fetal hepatic protein expression of translationally regulated Hsp40 and Hsp90 was also grossly increased among offspring of maternal high-fat diet animals when compared with controls (Fig. 4b).
Figure 4.

Quantification of fetal hepatic (a) mRNA expression of Hsp27 and Hsp70 and (b) Hsp 40 and Hsp90 protein expression in the primate under conditions of a maternal high-fat diet. (A) Real-time RT-PCR estimate of relative expression of Hsp27 and Hsp70 (ΔCT RNA was isolated from fetal hepatic liver under conditions of control (n=9) and high-fat (n=17) maternal diet in years 1 and 2, and quantification of fetal hepatic mRNA expression is shown as fold mRNA (mean±S.E.M.) expression relative following normalization ΔΔCT). Data are representative of a minimum of three separate experiments. (B) Translational expression of fetal hepatic Hsp40 and Hsp90. Quantification of 20 μg cytosolic protein was performed by western blotting following normalization. Data are representative of a minimum of three separate experiments.
Discussion
According to the fetal or developmental origins of adult disease hypothesis, perturbations in the gestational milieu influence the development of diseases later in life through the static reprograming of gene expression via alterations in chromatin infrastructure. We have previously shown that uteroplacental insufficiency induced through bilateral uterine artery ligation of the pregnant rat dam results in asymmetrical IUGR and, similar to the human, causes demonstrable postnatal disease; these alterations are associated with modifications of the fetal epigenome (changes in postnatal gene expression; Fu et al. 2004, 2006, MacLennan et al. 2004, Ke et al. 2005). Prior to the work performed herein, a number of crucial pieces of evidence integral to our understanding of Barker’s hypothesis as it relates to human disease were missing. First, modifications of the primate fetal epigenome in response to perturbations in the gestational milieu were assumed but unproven. This is of teleological importance, as the physiologic stress ‘felt’ by a fetus may differ in animals designed to carry a singleton gestation rather than litter; the fetal epigenome would be anticipated to modify accordingly. Secondly, it was unknown whether nutritional overabundance of epidemic proportions (e.g., maternal obesity) would profoundly modify fetal chromatin structure similar to that observed in the models of nutritional constraint.
We have demonstrated that a maternal high-fat diet results in lysine site-specific acetyl modifications of fetal hepatic chromatin structure (Fig. 1); the alterations in the expression and function of related key components of the epigenetic machinery accompany these modifications (Fig. 2). Taken together, our observations imply significance for several reasons. First, these data are a novel demonstration that the fetal primate epigenome is modified in response to a clinically relevant and evolutionarily recent in utero nutritional stress: a caloric-dense high-fat maternal diet leading to obesity. Secondly, at a molecular level, we observe that the fetal chromatin structure is not globally altered but rather modified via site-specific alterations in H3 acetylation (Fig. 1a and c).
We find it of interest that we did not observe global alterations in histone marks but rather observed lysine-specific punctuate modifications (i.e., significant alterations in H3K14 but not H3K9 nor and H3 methylation demarcation; Fig. 1). We hypothesize that this may be due to the importance of maintaining site specificity in the fetal primate epigenome. It is possible that fetal hepatic chromatin resides predominantly as inert H3K18 hetero-chromatin, and is reliant upon stress-induced deimination or demethylation for recruitment of histone acetylases to ultimately render chromatin accessible for transcription (Kasten et al. 2004, Pokholok et al. 2005, Shogren-Knaak et al. 2006, Yan & Boyd 2006, Guenther et al. 2007). Following gene activation, resumption of a repressed methylated state would employ HDAC-dependent recruitment of the methyltransferases to provide the methylation ‘marks’ integral to the control of epigenetic events akin to that recently observed in the rodent models on the developmental origins of disease (Lillycrop et al. 2007). Our observation that an ordered change in the ratio of modified acetyl variants (Fig. 1c) occurs under conditions associated with a marked decrease in HDAC1 expression and function alongside relatively minor alterations in site-specific methylation is certainly consistent with this notion. Moreover, organisms are classically assumed to adapt to any given fetal stress over time with a patterned, dichotomous site-specific histone modification (i.e., AcH3K14 hyperacetylation and H3K27me2me3), which is integral to the regulation of gene-specific transcription. However, in the face of a de novo evolutionary event (nutritional abundance), site-specific acetylation modifications of the histone code likely enable the regulation of genes integral to glucose and lipid homeostasis. In this fashion, modification of the epigenome is poised to occur via heritable site-specific covalent modifications rather than a potentially disastrous global modification of fetal chromatin structure (Lillycrop et al. 2007).
Acetylation of core histones on target promoters has been closely linked to transcriptional gene activation in both lower and higher eukaryotes (Li 2002, Kasten et al. 2004, Kurdiastani et al. 2004, Bernstein et al. 2005, Pokholok et al. 2005, Roh et al. 2005, Shogren-Knaak et al. 2006, Guenther et al. 2007). We utilized these observations of others alongside our findings presented in Figs 1 and 2 in developing our experimental design for ChIP. We anticipated that ChIP with an anti-AcH3K14 antibody would reveal a profile potentially consistent with alterations in gene-specific transcription, as well as identify the nature of ‘reprogramed’ genes potentially integral to the anticipated postnatal phenotype. However, it bears comment that our data and their interpretations are intrinsically limited by the current technology and available reagents for primate research. Given the absence of a macaque-specific microarray, whole genome profiling experiments are not feasible. Thus, definitive conclusions regarding the role of H3K14 acetylation with respect to altered gene expression per se cannot be discerned from our data.
Nevertheless, observed modest but not statistically significant alterations of fetal hepatic expression of GPT2 and DNAJA2 (Fig. 3) are consistent with corroborative evidence from microarray gene expression analysis in this same non-human primate model (Bishop et al. 2008), alongside murine models of obesity (Kim et al. 2004). Using similar microarray approaches in adult long-term high-fat intake-induced obesity murine models, others have observed an integral role of key Hsps (i.e., Hsp27, Hsp40, Hsp70, and Hsp90) and their co-chaperones (i.e., DNAJA2) along the IRS-1-inducible kinase cascades (Kim et al. 2004). With our initial demonstration of the perturbation of these pathways in utero following maternal high-fat diet exposure, it raises the possibility that transcriptional adaptation of Hsps and co-chaperones is similarly involved in the establishment and maintenance of the obese phenotype during the developmental and postnatal life. Our data presented herein recognize the potential contribution of gene-specific epigenetic modifications to these molecular adaptations.
Our identification of significant altered expression of Npas2 alongside Rdh12 as reprogramable genes integral in the temporal coordination of feeding behavior with peripheral metabolism in utero is of further dual interest (Simon et al. 1995, Ripperger & Schibler 2006). Circadian rhythms allow for the appropriate temporal synchronization (or entrainment) of physiology and behavior, thereby optimizing the efficiency of biological systems for the enablement of metabolic homeostasis. Npas2-deficient mice with an inability to adapt to food restriction are regarded as totally reliant on light/dark differentiation in the suprachiasmatic nucleus for circadian behavior (Dudley et al. 2003). We observe a down-modulation of fetal Npas2 expression in response to a maternal caloric-dense diet, implying a potential postnatal dysregulation of the normal hepatic entrainment response. Other investigators have recently shown that murine hepatocytes employ AcH3K9, H3K4me3, and H3K9me2 in regulating diurnal transcriptional oscillation of the Clock family members; this occurs via multiple E-box transcriptional elements in the Dbp locus (Ripperger & Schibler 2006). Our observed concomitant upregulation of Rdh12 is of heightened interest, as its family of short chain alcohol dehydrogenases similarly contains E-box motifs (Simon et al. 1995).
In summary, our data illustrate the capacity for a caloric-dense high-fat maternal diet to alter the fetal primate epigenome in an acetyl site-specific manner on the H3 tail. Such site-specific ‘permissive’ or ‘restrictive’ modifications of the histone code likely associate with the reprogramed expression of fetal genes. These in turn potentially favor the presumptive obese postnatal phenotype in an anticipatory fashion. Collectively, our observations lend a potential molecular basis to the developmental origins of adult disease in primates.
Acknowledgments
NIDDK 1R01DK079219-01 (K G) and NICHD 1R01DK R01DK080558-01 (R H L).
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
References
- Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436:1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–174. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang S, Wei G, Chepelev I, Zhao K. High resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bhargava SK, Sachdev HS, Fall CH, Osmond C, Lakshmy R, Barker DJ, Biswas SK, Ramji S, Prabhakaran D, Reddy KS. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. New England Journal of Medicine. 2004;350:865–875. doi: 10.1056/NEJMoa035698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JM, McCurdy CE, Williams SM, Smith MS, Friedman JE, Grove KL. Chronic maternal high fat diet triggers fetal hepatic reprogramming and fatty liver in the nonhuman primate. FASEB -Journal. 2008 doi: 10.1172/JCI32661. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. British Journal of Nutrition. 2007;97:435–439. doi: 10.1017/S0007114507352392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003;301:379–383. doi: 10.1126/science.1082795. [DOI] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilheto J, Osmond C, Barker DJP. Early growth and coronary heart disease in later life: longitudinal study. BMJ. 2001;322:949–953. doi: 10.1136/bmj.322.7292.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AE, Coakely EH, Must A, Spadano JL, Laird N, Dietz WH, Rimm E, Colditz GA. Impact of overweight on the risk of developing common chronic diseases during a 10 year period. Archives of Internal Medicine. 2001;161:1581–1586. doi: 10.1001/archinte.161.13.1581. [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Ruhm CJ, Kosa KM. Economic causes and consequences of obesity. Annual Review of Public Health. 2005;26:239–257. doi: 10.1146/annurev.publhealth.26.021304.144628. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Yu X, Wang L, Callaway CW, Lane RH. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiological Genomics. 2004;20:108–116. doi: 10.1152/physiolgenomics.00175.2004. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Yu X, Callaway CW, Lane RH. Growth retardation alters the epigenetic characteristics of hepatic dual specificity phosphatase 5. FASEB Journal. 2006;20:2127–2129. doi: 10.1096/fj.06-6179fje. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, Burdge GC, Hanson MA. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. PNAS. 2007;104:12796–12800. doi: 10.1073/pnas.0705667104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kasten M, Szerlong H, Erdjument-Bromage H, Tempst P, Werner M, Cairns BR. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys 14. EMBO Journal. 2004;23:1348–1359. doi: 10.1038/sj.emboj.7600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke X, McKnight RA, Wang ZM, Yu X, Wang L, Callaway CW, Albertine KH, Lane RH. Nonresponsiveness of cerebral p53-MDM2 functional circuit in newborn rat pups rendered IUGR via uteroplacental insufficiency. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2005;288:R1038–R1045. doi: 10.1152/ajpregu.00701.2004. [DOI] [PubMed] [Google Scholar]
- Kim S, Sohn I, Ahn J-I, Lee K-H, Lee YS. Hepatic gene expression profiles in a long-term high-fat diet-induced obesity mouse model. Gene. 2004;340:99–109. doi: 10.1016/j.gene.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Klebanoff MA, Meirik O, Berendes HW. Second-generation consequences of small-for-dates birth. Pediatrics. 1989;84:243–247. [PubMed] [Google Scholar]
- Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- Kurdiastani SK, Tavazoi S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to gluococorticoids and intrauterine growth retardation, and disturbs the hypothalamic–pituatary adrenal axis in the newborn rat. Endocrinology. 2001;142:1692–1702. doi: 10.1210/endo.142.5.8139. [DOI] [PubMed] [Google Scholar]
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature Reviews Genetics. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferace-1 expression is involved in impaired DNA methylation changes in histone modifications. British Journal of Nutrition. 2007;97:1064–1073. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan NK, James SJ, Melnyk S, Piroozi A, Jernigan S, Hsu JL, Janke SM, Pham TD, Lane RH. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiological Genomics. 2004;18:43–50. doi: 10.1152/physiolgenomics.00042.2004. [DOI] [PubMed] [Google Scholar]
- Martens JH, OSullivan RJ, Braunscheig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO Journal. 2005;24:800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, Nair A, Mellor J. Dynamic lysine methylation on histone H3 defines the regulatory phase of gene transcription. Molecular Cell. 2005;18:723–734. doi: 10.1016/j.molcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. Journal of the American Medical Association. 1999;282:1523–1529. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- Pastewka JV, Reed R, Ness AT, Peacick AC. An improved haptoglobin subtyping procedure using polyacrylimide gel electrophoresis. Analytical Biochemistry. 1973;51:152–162. doi: 10.1016/0003-2697(73)90463-6. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison DR, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Popkin BM, Gordon-Larsen P. The nutrition transition: worldwide obesity dynamics and their determinants. International Journal of Obesity. 2004;28:S2–S9. doi: 10.1038/sj.ijo.0802804. [DOI] [PubMed] [Google Scholar]
- Rennie KL, Jebb SA. Prevalence of obesity in Great Britain. Obesity Reviews. 2005;6:11–20. doi: 10.1111/j.1467-789X.2005.00164.x. [DOI] [PubMed] [Google Scholar]
- Rich-Edwards JW, Kleinman K, Michels KB, Stampfer MJ, Manson JE, Rexrode KM, Hibert EN, Willett WC. Longitudinal study of birth weight and adult body mass index in predicting coronary heart disease and stroke in women. BMJ. 2005;330:1115–1118. doi: 10.1136/bmj.38434.629630.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nature Genetics. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes and Development. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouszrides T. Active genes are trimethylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M, Peng L, Puga A. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochimica et Biophysica Acta. 2007;1769:569–578. doi: 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishiii H, Sun JFM, Pazin MF, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Simon A, Hellman U, Wernstedt C, Eriksson U. The retinol pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short chain alcohol dehydrogenases. Journal of Biological Chemistry. 1995;270:1107–1112. [PubMed] [Google Scholar]
- Slyper AH. The pediatric obesity epidemic: causes and controversies. Journal of Clinical Endocrinology and Metabolism. 2004;89:2540–2547. doi: 10.1210/jc.2003-031449. [DOI] [PubMed] [Google Scholar]
- Speiser PW, Rudolf MC, Anhalt H, Camacho-Hubner C, Chiarelli F, Eliakim A, Freemark M, Gruters A, Hershkovitz E, Iughetti L, et al. Childhood obesity. Journal of Clinical Endocrinology and Metabolism. 2005;90:1871–1887. doi: 10.1210/jc.2004-1389. [DOI] [PubMed] [Google Scholar]
- Stein CE, Fall CH, Kumaran K, Osmond D, Cox V, Barker DJ. Fetal growth and coronary heart disease in South India. Lancet. 1996;348:1269–1273. doi: 10.1016/s0140-6736(96)04547-3. [DOI] [PubMed] [Google Scholar]
- Strauss RS, Pollack HA. Epidemic increase in childhood overweight, 1986–1998. Journal of the American Medical Association. 2001;286:2845–2848. doi: 10.1001/jama.286.22.2845. [DOI] [PubMed] [Google Scholar]
- Yan C, Boyd DD. Histone H3 acetylation and H3K4 methylation define distinct chromatin regions permissive for transgene expression. Molecular and Cellular Biology. 2006;26:6357–6371. doi: 10.1128/MCB.00311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]