

Abstract
Background
Pathological angiogenesis is an intrinsic component of chronic intestinal inflammation, which results in remodeling and expansion of the gut microvascular bed. Endoglin is essential for endothelial cell function and physiological angiogenesis. In this study, we investigated its potential role in the regulation of inflammation by testing the response of Endoglin heterozygous (Eng+/-) mice to experimental colitis.
Methods
C57BL/6 Eng+/- and littermate control mice drank water supplemented with 3% dextran sulfate sodium (DSS) for 5 days and were monitored for up to 26 days for clinical signs of colitis. Inflammation, crypt damage and angiogenic index were scored on histological sections of distal colon. Levels of the vascular endothelial growth factor (VEGF) and angiopoietins were measured by real time polymerase chain reaction, ELISA and/or Western blots. Vascular permeability was assessed using Evans Blue.
Results
Eng+/- and control mice developed acute colitis, which peaked at day 9. While control mice recovered by days 19-26, Eng+/- mice progressed to chronic colitis and showed numerous vascular protrusions penetrating into the serosa of the inflamed distal colon. Prior to DSS induction, VEGF levels and vascular permeability were higher in the distal colon of Eng+/- mice, while angiopoietin 1 and 2 levels were unchanged. In the chronic phase of colitis, VEGF levels were increased in both groups of mice and remained significantly higher in the Eng+/- mice.
Conclusions
Higher VEGF levels and increased vascular permeability in the distal colon may predispose Eng+/- mice to progress to chronic and persistent bowel inflammation, associated with pathological angiogenesis.
Keywords: endoglin, VEGF, angiogenesis, inflammation, inflammatory bowel disease
INTRODUCTION
The pathogenesis of inflammatory bowel disease (IBD) involves several cell types including endothelial cells, which contribute to multiple aspects of intestinal inflammation. The vascular endothelium via up-regulation of adhesion molecules and secretion of chemokines is responsible for the recruitment of leukocytes to the site of inflammation.1 In turn, the endothelium responds to the pro-angiogenic cytokines produced by the infiltrating cells in a process referred to as immune-driven angiogenesis.2,3 The mucosal microcirculation becomes angiogenically active and contributes to the development of new vessels, which then through their functional activation recruit more immune cells and sustain chronic inflammation.4,5 The extent of vascular involvement is correlated with disease severity and chronically inflamed areas of the gut are characterized by extensive angiogenesis, particularly in the bowel wall and serosal surfaces.6-8
Recent studies indicate that the vascular endothelial growth factor, VEGF-A, (referred to as VEGF), is elevated in colonic samples of Crohn’s disease (CD) and ulcerative colitis (UC) patients, relative to healthy controls.9 Overexpression of VEGF in mice with colitis experimentally induced with dextran sulfate sodium (DSS) worsened their disease and increased mucosal angiogenesis.9 VEGF is the major angiogenic and vascular permeability factor and is involved in several chronic inflammatory disorders.10-13 VEGF has a pro-inflammatory role in IBD pathogenesis but also drives the angiogenesis associated with intestinal inflammation and particularly with the chronic phase of disease. Angiopoietins (Ang) 1 and 2, both acting via the Tie-2 receptor, are implicated in the regulation of angiogenesis and their involvement in IBD has been reported in few studies. Serum levels of Ang2 were slightly elevated in patients with CD and UC relative to matched controls, and correlated with disease activity.14 A second study supported these observations in CD patients, and observed lower levels of Ang1 relative to healthy controls.15 A recent report suggested that the Ang/Tie pathway may play a role in the progression of UC.16 Ang1 helps in stabilizing normal vessels while Ang2 is expressed at sites of vascular remodeling where it blocks the effect of Ang1. In the presence of VEGF, Ang2 can induce an angiogenic response. Therefore an imbalance in Ang1 and Ang2 with a net gain in Ang2 could be associated with pathological angiogenesis in IBD.
The normal outcome of an acute inflammatory response is resolution and repair of the damaged tissue. Transforming growth factor-beta (TGF-β̣ is essential for wound healing and tissue repair and implicated in the resolution of inflammation. Mice deficient in TGF-β1 die of multifocal inflammatory disease by 3 weeks of age.17,18 TGF-β is a multipotent cytokine, which affects cellular proliferation and migration in several cell types and organs including the vascular endothelium. It inhibits endothelial cell proliferation but can also regulate VEGF production.19 There is cross-talk between VEGF and TGF-β,20 and dysregulated TGF-β pathways can therefore affect angiogenesis and vascular permeability. Endoglin is an endothelial specific co-receptor for TGF-β implicated in the maintenance of vascular integrity and regulation of vascular tone.21,22 Mutations in the ENDOGLIN gene lead to hereditary hemorrhagic telangiectasia type 1 (HHT1), an autosomoal dominant vascular dysplasia characterized by arteriovenous malformations and altered angiogenesis in multiple organs including the gut.23 Mutations in the ALK1 gene, which codes for an endothelial cell specific TGF-β type I receptor that binds endoglin, lead to HHT2 which is often associated with gastrointestinal complications.23
To assess the contribution of endoglin to angiogenesis in the context of inflammation, we investigated the response of Endoglin heterozygous (Eng+/-) mice to an experimental model of IBD. We also assessed the potential role of VEGF, Ang1 and Ang2 in the angiogenic response of these mice. Eng+/- mice were shown to express higher levels of VEGF in the colon and increased vascular permeability compared to control mice, while Ang1 and Ang2 were unchanged. When subjected to a single course of the chemical irritant DSS, the Eng+/- mice progressed to chronic gut inflammation while the control mice recovered after the acute phase of disease. Chronic inflammation in the Eng+/- mice was accompanied by a further increase in VEGF and extensive angiogenesis. The current study reveals that the Eng+/- mice represent a novel model of chronic IBD, where TGF-β mediated dysregulation of VEGF and vascular permeability leads to chronic inflammation associated with pathological angiogenesis.
MATERIALS AND METHODS
Mice
Congenic 14-15 week old Eng+/− mice on the C57BL/6 background (at generations 20-22) and their control littermates (Eng+/+) were used. Eng+/− mice were first generated by homologous recombination using embryonic stem cells of 129/Ola origin, and then backcrossed to C57BL/6.24 Mice were kept in ventilated racks in a specific pathogen-free facility. All protocols were approved by the Animal Care Committee, at the Hospital for Sick Children, in accordance with the Canadian Council on Animal Care. The genotype of each mouse was assessed at weaning time by β-galactosidase staining of an ear punch and subsequently confirmed by polymerase chain reaction (PCR) using tail DNA.25
DSS-induced colitis
In all experiments, mice drank acidified water supplemented with 3% DSS (DSS, m.w. 36,000-50,000, MP Biomedicals, Illkirch, France) for 5 days, starting at day zero, and were then returned to normal water on day 5.26 Mice were examined daily for up to 26 days for body weight, water and food intake, activity, fur appearance, and diarrhea was estimated with slight modifications of Ohkawara’s procedure.26 Diarrhea was scored using a 0-to-4 scale: 0- normal, 1- slightly loose feces, 2- loose feces, 3- semi-liquid stool, and 4- liquid stool and fecal occult blood (by guaiac paper, ColoScreen Hemocult kit, Helena Labs, Beaumont, TX). At time of sacrifice, mice were anesthetized with ketamine (100mg/kg i.p.) and xylazine (10 mg/kg i.p.), perfused with 10 mL of phosphate-buffered saline (PBS) through the left ventricle, and the colon was carefully removed and its length measured. Colonic samples were prepared for histopathology or snap frozen in liquid N2 and stored at -80°C for subsequent protein and RNA extraction.
Histology
Cecum and colon (transversal and distal) samples were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned longitudinally and stained with hematoxylin and eosin. Sections were evaluated and scored for inflammation, regeneration, and crypt damage (adapted from Dieleman, 1998)27 as follows: a) inflammation assessed by leukocyte infiltration [0, none; 1, slight; 2, moderate; 3, severe]; b) epithelium regeneration [0, complete or normal tissue; 1, almost complete; 2, some crypt depletion; 3, surface epithelium not intact; 4, no tissue repair]; c) crypt damage [0, none; 1, basal 1/3 damaged; 2, basal 2/3 damaged; 3, whole crypt damaged but surface epithelium still intact; 4, complete loss of crypt and epithelium]; d) percent involvement [1 for 1-25%, 2 for 26-50%, 3 for 51-75%, and 4 for 76-100%]. The a), b) and c) scores were then multiplied by the percent involvement score and their sum estimated to derive the total colitis histological score.
Other colon sections were stained with Movat pentachrome, which marks particularly well blood vessel elastin. On these slides, the angiogenic score, defined in our system by the number of vascular protrusions (which include one to several vessels) arising in the lamina propria and penetrating through the muscular layer and into the serosa, was assessed in distal colon samples. The inner area of all vessels (of diameter < 60 μm) present in submucosal, muscular and serosal layers at day 19 was measured in cecum, transversal and distal colon using the OpenLab software.
Vascular permeability (VP)
Mice were injected with 100 μL of 2% Evans blue dye (Sigma-Aldrich Canada Ltd, Oakville, ON) in PBS, and after 40 minutes anesthetized, bled by heart puncture (and plasma prepared) and perfused through the heart with PBS.28 The colon was removed and separated from the cecum, and cut into distal and transversal portions. Samples were fixed in formamide for 24h at 70°C and optical density (O.D.) of the released dye measured at 620 nm and 740 nm. The following formula was used to correct for heme pigment contamination: O.D. 620 (corrected) = O.D 620-(1.326 × O.D740 + 0.030). The concentration of Evans blue was calculated per gram of tissue and expressed as a tissue/plasma ratio.29
Real time PCR for VEGF, Ang1 and Ang2 mRNA levels
Total RNA from approximately 30-40 mg of frozen distal colon was extracted using TRIzol reagent (Invitrogen Canada Inc., Burlington, ON) according to manufacturer’s instructions. The RNA extracts were treated with amplification grade DNase I (Invitrogen), and cDNA was generated using the Moloney Murine Leukemia Virus reverse transcriptase (RT), Superscript II (Invitrogen). For each sample, 10ng of cDNA was subjected to real-time PCR, using pre-formulated TaqMan® Gene Expression Assays (Applied Biosystems, CA) for VEGF-A (Mm00437306_m1), Ang1 (Mm00456503_m1), Ang2 (Mm00545822_m1) and GAPDH (Mm99999915_g1). Samples were run on the Standard 7500 Real Time PCR system from Applied Biosystems. VEGF, Ang1 and Ang2 mRNA levels were normalized to GAPDH levels.
Tissue VEGF protein levels determined by ELISA
Distal colonic tissue was homogenized in lysis buffer (50mM Tris-HCl pH 7.4, 100 mM NaCl, 1mM EDTA, 1% Triton X-100), with complete protease inhibitors (Roche Diagnostics GmbH, Mannheim, Germany). After rotation at 4°C for 30 min, the lysates were centrifuged at 13,000 × g for 15 min and the supernatants collected. Total protein concentration was measured using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Mouse VEGF Immunoassay (Quantikine, R&D Systems, Minneapolis, MN) was used for quantitative determination of VEGF-A protein in distal colonic tissue according to the manufacturer’s protocol and using approximately 200 μg of proteins per sample.
Western blot analysis of Endoglin, VEGF and Ang2
Distal colonic lysates prepared as above, were incubated in Laemmli buffer at 95°C for 5 min and electrophoresed on 4-12% gradient SDS/PAGE gels (Invitrogen, Canada Inc., Burlington, ON). Fractionated proteins were electro-transferred and the PDVF membranes blocked with 5% milk in TBS-T (20 mM Tris, pH 7.6, 137 mM NaCl, 0.1 % Tween-20) for 1 h at 23°C. The blots were incubated at 4°C overnight with commercially available antibodies: CD105/Endoglin, rat IgG2a clone MJ7/18, 1:500 dilution, (Southern Biotech, Birmingham, AL); VEGF, humanized mouse IgG2a clone B20-4.1.1, 1:10,000 (Genentech Inc., San Francisco, CA); goat anti-Ang2, 1:500 (Santa Cruz Biotechnology Inc). After washing with TBS-T for 30 min, the blots were incubated with appropriate secondary antibodies conjugated with horseradish peroxidase (goat anti-rat and sheep anti-mouse IgG, GE Healthcare, Little Chalfont, England; or donkey anti-goat IgG, Santa Cruz Biotechnology Inc; 1:10,000 dilution in TBS-T containing 3% milk) for 60 min at 23°C, and washed with TBS-T. The enhanced chemiluminescence ECL reagent (Perkin Elmer, Shelton, CN) was used for detection. The membranes were stripped and probed with antibodies to β-actin (Sigma Aldrich; dilution 1:10,000).
Statistical Analysis
Data were evaluated by one- or two-way analysis of variance (ANOVA) with multiple comparisons obtained by the Newman-Keuls test, and corrected for repeated measures when appropriate. Survival curves were analyzed using the Kaplan-Meier method and log-rank test. The histological colitis, inflammation, and angiogenic scores were analyzed using the non-parametric Wilcoxon rank test. Statistical significance was accepted at P < 0.05. Statistics including regression and correlation analyses were performed using Primer of Biostatistics (Stanton Glantz Program) and SAS software programs. Data are presented as means ± SEM.
RESULTS
Eng+/− mice show decreased survival and more severe colitis than control mice when exposed to DSS
Mice receiving DSS orally can develop acute or chronic colitis resembling ulcerative colitis30 (UC) and associated with weight loss, shortening of the gut, mucosal ulcers and leukocyte infiltration.31 The C57BL/6 strain, background for the Eng+/- congenic mice, is a good responder to a single course of the DSS chemical irritant and develops a robust disease, most severe in the distal colon and highly reproducible.32 We compared Eng+/- congenic mice and littermate controls for their response to DSS. Mice drank water containing 3% DSS for 5 days and then normal water for the rest of the experiment. They were observed daily for water and food intake and signs of colitis for up to 26 days. Survival was significantly less in the Eng+/- than in the Eng+/+ group (62% versus 75%) (Fig. 1A). Weight loss began at day 5 and reached 25% by day 8 in the Eng+/- mice compared to 20% in the Eng+/+ mice (Fig. 1B). Mice began to regain weight by day 10 in both groups. By day 18, the weight of the Eng+/+ DSS-treated group was no longer different from the untreated group while that of the DSS-treated Eng+/- group was still significantly different from control at day 26. Fecal blood (Fig. 1C) and diarrhea (Fig. 1D) were most pronounced at day 7 and remained elevated in both groups but were significantly higher in the Eng+/- group throughout the course of disease. Upon sacrifice, colon length was measured and shown to be shorter after DSS treatment in both groups of mice on days 5, 9 and 19 (Fig. 2). A greater reduction in colon length was observed in Eng+/− than in Eng+/+ mice at days 9, 19 and 26.
FIGURE 1.
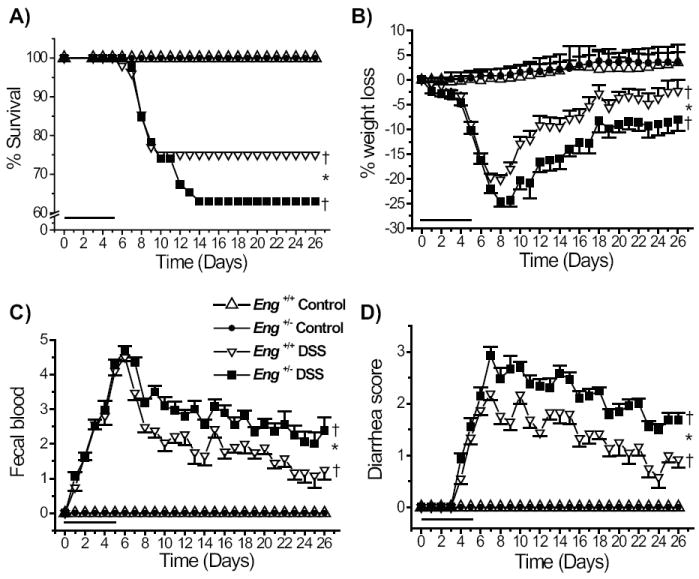
Reduced survival and more severe colitis in DSS-treated Eng+/− compared to Eng+/+ mice. All mice drank water containing 3% DSS for 5 days (as indicated by the bar) and then normal water; they were monitored daily for weight changes and clinical signs for up to 26 days. A) Survival of the DSS-treated Eng+/− mice was lower than for the DSS-treated Eng+/+ mice. Kaplan-Meier plots and log-rank tests were used. The DSS-treated groups (N = 46) had reduced survival compared to the respective untreated groups (N = 10). †P < 0.05 vs. corresponding control untreated group; *P < 0.05 vs. DSS-treated Eng+/+ mice. B) Greater weight loss in DSS-treated Eng+/− mice than in DSS-treated Eng+/+ mice (*P < 0.05); both DSS-treated groups lost weight starting at day 5 compared to control groups (†P < 0.05). C) Fecal blood was first noted in the DSS-treated mice at day 2, was maximal at day 7 and remained higher in the Eng+/− group (*P < 0.05) throughout the whole experiment. D) Diarrhea was first observed at day 4, maximal at day 7 and remained higher in the Eng+/− vs. Eng+/+ group (*P < 0.05).
FIGURE 2.
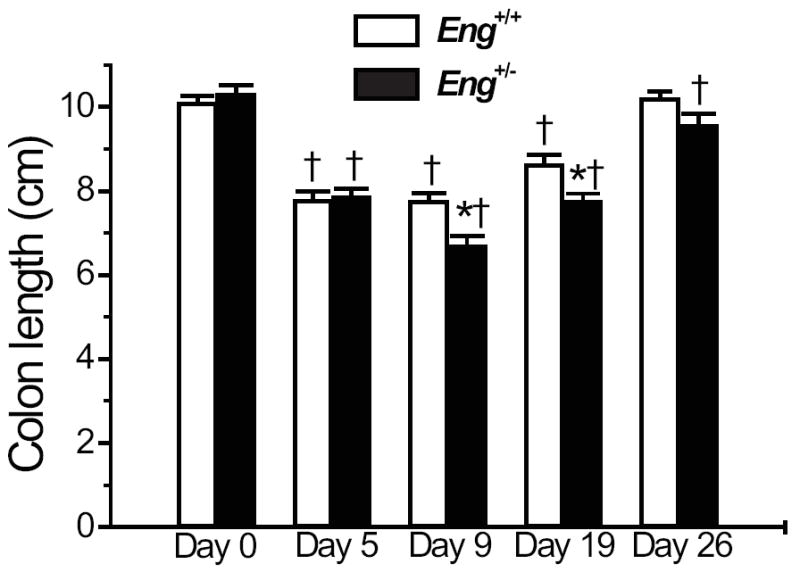
Colon length is further reduced in DSS-treated Eng+/− mice. Mice were treated as described in FIGURE 1 and sacrificed at the time points indicated. Colon length was shorter after DSS treatment in both groups of mice on days 5, 9 and 19 (†P < 0.05 relative to corresponding untreated group). The DSS-treated Eng+/− mice showed further shortening of the colon at days 9 and 19 (*P < 0.05 vs. DSS-treated Eng+/+ mice).
Eng+/− mice progress to chronic disease while Eng+/+ mice recover after the acute phase of colitis
Histological examination of isolated colonic tissue was performed for untreated mice (control) and after DSS treatment on days 5, 9, 19 and 26. Initial analysis revealed that the distal colon was most affected followed by cecum and that the transversal colon showed less signs of disease. Distal colon sections are illustrated (Fig. 3). No difference was observed between Eng+/+ and Eng+/− mice prior to DSS treatment. The peak of inflammation was observed at day 9 in both groups of mice: leukocyte infiltration was massive and crypt damage was highly visible in the submucosa. By day 19, signs of resolution were noted in the Eng+/+ mice as leukocyte infiltration diminished and crypt regeneration began. However, Eng+/− mice showed persistent infiltration and crypt damage (Fig. 3). By day 26, Eng+/+ mice were almost fully recovered while Eng+/− mice displayed residual infiltration and crypt damage and widening of the serosal layer, characteristic of chronic inflammation.
FIGURE 3.
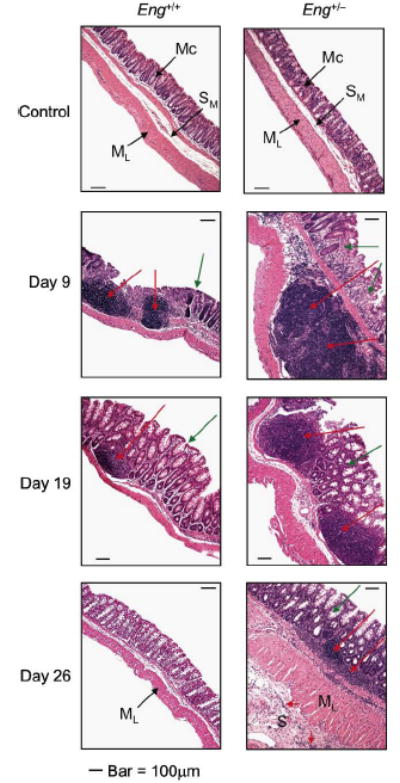
DSS-treated Eng+/− mice progress to a chronic phase of disease while DSS-treated Eng+/+ mice recover following the acute phase. Mice were treated as described in FIGURE 1 and sacrificed as indicated. Distal colon sections were stained with Hematoxylin and Eosin. Normal mucosa (Mc), submucosa (SM) and muscle layer (ML) were very similar in control Eng+/+ and Eng+/− mice prior to DSS treatment. At day 9, maximum leukocyte infiltration (red arrows) and crypt damage (green arrows) were observed in the DSS-treated Eng+/+ and Eng+/− mice. At day 19, Eng+/+ mice showed reduced leukocyte infiltration and crypt damage compared to day 9 while Eng+/− mice had persistent inflammation. By day 26, Eng+/+ mice had recovered while Eng+/− mice showed only partial epithelial regeneration, some residual infiltration even in the serosa (S) and enlarged ML and S layers, characteristic of chronic inflammation. Bar = 100 μm.
Extensive angiogenesis is associated with the chronic phase of inflammation in Eng+/- mice
Inspection of sections of distal colon, stained with Movat pentachrome to highlight the blood vessels, revealed that the microvasculature had undergone extensive angiogenesis, particularly in the DSS-treated Eng+/- mice. Vessels originating in the submucosa and penetrating through the lamina muscularis propria and into the serosa were observed in both DSS-treated groups at days 5 and 9 (Fig. 4). The vascular protrusions (containing one to several vessels) were still numerous in the Eng+/- groups at days 19 and 26 while barely detectable in the Eng+/+ groups (Fig. 4). Thus the Eng+/- mice show pathological angiogenesis correlating with persistent chronic colitis, while control mice recover.
FIGURE 4.
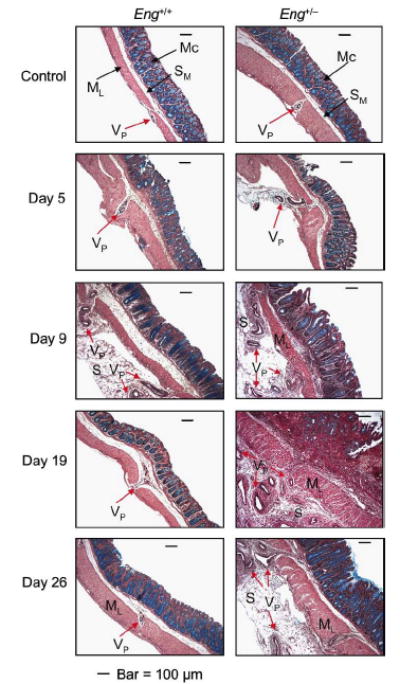
Mucosal microvessels undergo more extensive angiogenesis in the distal colon of the DSS-treated Eng+/− mice. Mice were treated as described in FIGURE 1 and sacrificed as indicated. Distal colon sections were stained with Movat pentachrome to highlight the blood vessels. In the control groups, few vascular protrusions with one or more vessels (VP) from the submucosa (SM) through the muscle layer (ML) were observed. Vessels of increasing size and in larger numbers protruded from SM through ML and into the serosa (S), starting from day 5 and peaking at day 9 in the Eng+/+ mice; a recovery phase followed with VP showing a near normal pattern by days 19-26. In the Eng+/− mice, the vascular remodeling persisted beyond day 9 with a large number of VP lodging into the serosa at days 19-26. Bar = 100 μm.
To determine potential correlations between colitis, inflammation and the number of vascular protrusions, we compared the total colitis histological score, the inflammation score and the number of vascular protrusions from submucosa into serosa. The total colitis histological score, which integrates inflammation, lack of regeneration, and crypt damage scores, was maximal between days 5-9 in both groups; at days 19 and 26, it was significantly higher in DSS-treated Eng+/− than in Eng+/+ mice (Fig. 5A). The inflammation score based on leukocyte infiltration and the percentage of colonic involvement was maximal at day 9 in both groups and was also significantly higher in the Eng+/− groups at days 19 and 26 (Fig. 5B).
FIGURE 5.
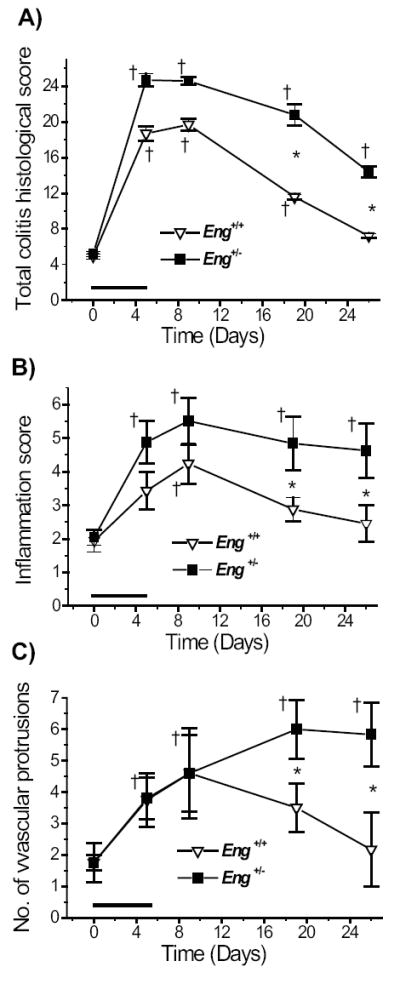
Colitis, inflammation and angiogenesis scores are higher in DSS-treated Eng+/− than Eng+/+ mice. Tissue sections were scored to quantify the extent of inflammation and angiogenesis. A) Total colitis histological score was calculated from the sum of the inflammation, lack of regeneration, and crypt damage scores on hematoxilin and eosin stained sections as described in Methods. B) Inflammation score was based on leukocyte infiltration and percent gut involvement on the same sections. C) The total number of vascular protrusions (containing one to several vessels) arising from the submucosa and penetrating through the lamina muscularis and into the serosa was counted for the whole distal colon on Movat pentachrome stained sections. †P < 0.05 vs. corresponding control untreated group (at day 0); *P < 0.05 vs. DSS-treated Eng+/+ mice; N = 6 for the DSS-treated groups at all time points; N = 5 for the control groups. Data were analyzed using the non-parametric Wilcoxon rank test.
The total number of vascular protrusions emerging from the submucosa into the serosa was estimated for the whole distal colon (Fig. 5C). It increased in both groups of mice till day 9 in an identical manner, likely reflecting the angiogenesis associated with the acute phase of inflammation. After day 9, the number of protrusions decreased in the Eng+/+ mice returning to normal by day 26. However, in the Eng+/− mice, the number of vascular protrusions was highest at days 19-26 and associated with the chronic phase of inflammation.
Correlation analyses were performed using the data points shown in Fig. 5. The strongest correlation to emerge was between the inflammation score and the angiogenic score in Eng+/+ mice (r = 0.97) indicating that both processes are normal components of an acute inflammatory response. In the Eng+/− mice, a lower correlation value was observed between inflammation and angiogenesis (r = 0.77), which may be due to extensive pathological angiogenesis becoming the predominant process in the chronic phase of colitis at days 19-26.
The presence of abnormally dilated vessels in the distal colon and cecum was observed in DSS-treated Eng+/− mice during the chronic phase of colitis. A representative image is shown in Fig. 6A, which reveals enlarged vessels in the expanding serosal layer at day 19 in Eng+/− mice, while smaller vessels confined to the muscular layer were seen in the Eng+/+ mice. The inner area of all vessels present in colon sections at day 19 of colitis was measured and shown to be significantly greater in cecum and distal colon of Eng+/− vs. Eng+/+ mice (Fig. 6B).
FIGURE 6.
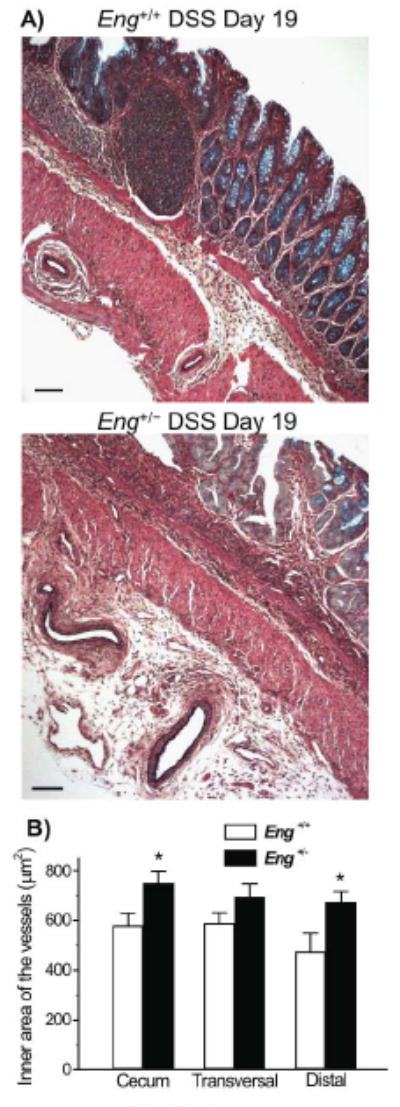
Enlarged vessels are observed in the colon of DSS-treated Eng+/− mice during the chronic phase of colitis. A) Representative images of Movat pentachrome stained distal colon sections showing dilated vessels in the serosa of Eng+/− mice. In Eng+/+ mice, vessels were smaller and found in the muscle layer. Bar =100 μm. B) The inner area of all vessels (<60 μm) present in the sections of cecum, transversal colon and distal colon was measured. Vessels were significantly larger in cecum and distal colon respectively when comparing Eng+/− and Eng+/+ mice (*P < 0.05; N=5).
Increased vascular permeability in Eng+/− mice
In order to assess if the progression to chronic inflammation in the Eng+/− mice was due to an intrinsic defect in the integrity of their vascular endothelium, we assessed vascular permeability (VP) in the colon and plasma of untreated Eng+/− and Eng+/+ mice using Evans blue. A significant increase in VP was observed in the cecum, transversal and distal colon of Eng+/− versus Eng+/+ mice, suggesting that the endothelium is compromised in Eng+/− mice (Fig. 7).
FIGURE 7.
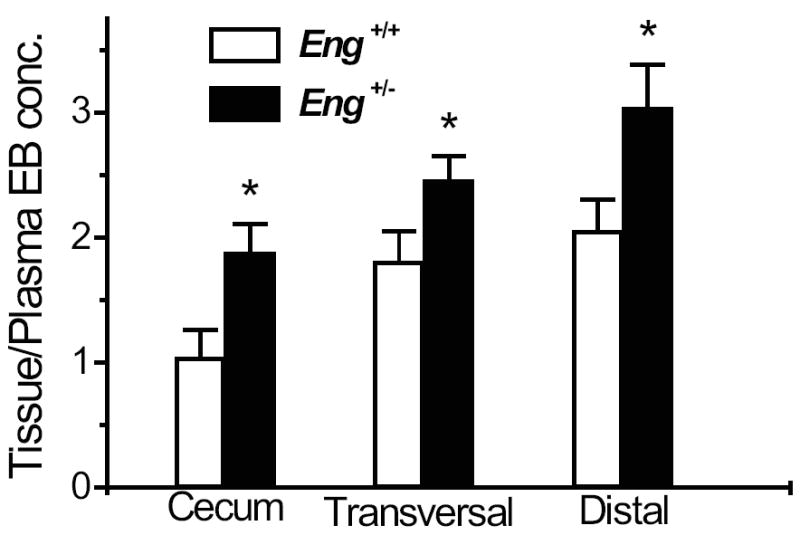
Increased vascular permeability in Eng+/− mice. Permeability was measured in non-DSS treated mice, using 2% Evans blue (EB) as described in Methods. The concentration of Evans blue was expressed per gram of tissue and relative to the plasma concentration. A significant increase in vascular permeability was observed in the cecum, transversal and distal colon of Eng+/− vs. Eng+/+ mice (*P < 0.05; N = 6-12).
Higher VEGF levels in distal colon of Eng+/− mice are further increased after DSS induction
We analyzed by Western blot the expression of endoglin and VEGF-A (VEGF164) in the distal colon, under basal conditions and on day 19 after DSS induction. Endoglin levels, estimated by quantifying the dimeric membrane protein of 170kDa, were reduced by half in the distal colon of Eng+/- relative to Eng+/+ mice, confirming the functional haploinsufficiency. Endoglin levels were not altered by the DSS treatment in either group (Fig. 8A).
FIGURE 8.
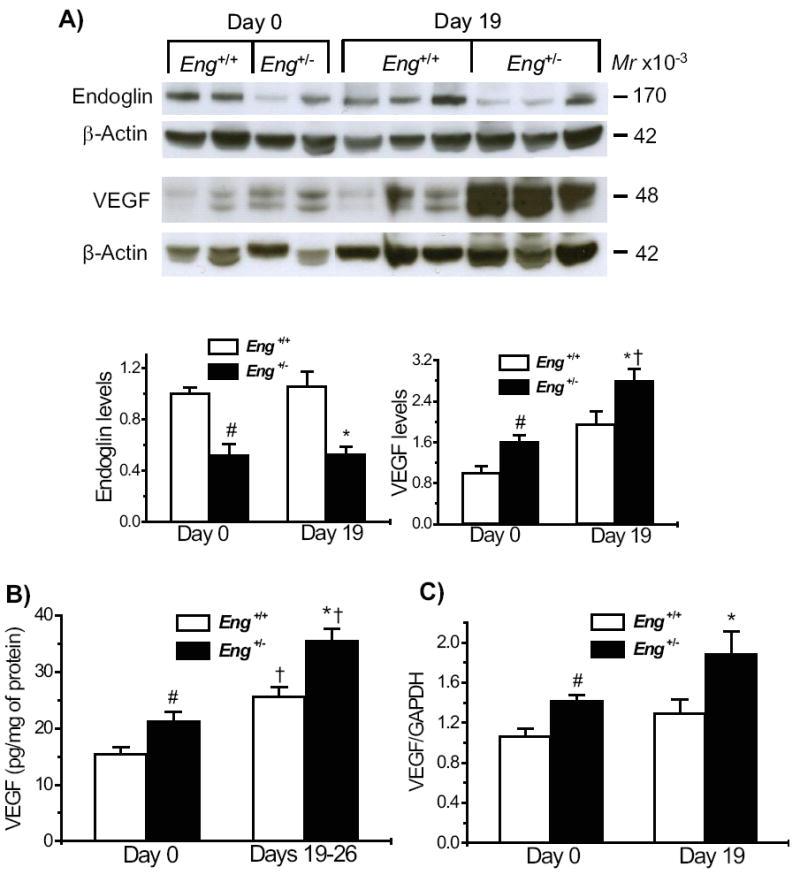
Increased VEGF-A levels in Eng+/− mice under basal conditions and after DSS-induced colitis. A) Endoglin and VEGF levels were estimated in colonic protein samples fractionated under non-reducing conditions, expressed relative to β-actin and normalized to the level of untreated Eng+/+ mice. Endoglin was significantly reduced in Eng+/− samples and no changes were observed following DSS induction. VEGF levels were increased in Eng+/− vs. Eng+/+ mice, prior to DSS induction, with a further increase at day 19. B) Protein levels of VEGF-A estimated by ELISA were higher in Eng+/− than Eng+/+ mice under basal conditions and in the chronic phase of colitis (Days 19-26). C) mRNA levels estimated by real time PCR were significantly higher in Eng+/− than Eng+/+ mice prior to treatment and on day 19, following DSS treatment. #P < 0.05 vs. Eng+/+ mice at day 0; #P < 0.05 vs. DSS-treated Eng+/+ mice; †P < 0.05 vs. corresponding day 0 values. In Western blots, N = 4-6 mice for endoglin and N = 5-6 for VEGF; in ELISA, N = 9-11 for all groups; in real time PCR analysis, N = 4-5 samples for all groups, with a minimum of 4 replicates per sample.
Intestinal VEGF was resolved as a dimeric doublet, at 48kDa, present in significantly higher amounts in the Eng+/- than Eng+/+ mice, under basal conditions. After DSS induction, both groups showed a significant increase relative to their day 0 levels; VEGF remained higher in the Eng+/- versus Eng+/+ group (Fig. 8A). All changes in VEGF protein levels were confirmed by the ELISA assay (Fig. 8B). Thus, VEGF expression was significantly increased by DSS treatment in both groups of mice, the highest level being associated with the extensive angiogenesis observed in the Eng+/- mice during the chronic phase of colitis.
VEGF mRNA present in colonic samples was estimated to ascertain if the increase seen in the Eng+/- groups was at the transcriptional level. VEGF levels were increased in Eng+/- mice relative to Eng+/+ mice before DSS induction and during the chronic phase of colitis (1.3 fold in both cases) (Fig. 8C).
Ang1 and Ang2 levels are unchanged in the distal colon of Eng+/+ mice
Ang1 and Ang2 can affect the angiogenic balance. We determined whether they were implicated in the Eng+/- model of IBD. Ang2 protein levels, measured by Western blot, were not different between Eng+/- and Eng+/+ mice, under basal conditions or on day 19 after DSS induction (Fig. 9A). No significant increase in colonic Ang2 was induced by DSS treatment in either Eng+/- or Eng+/+ mice. Ang2 mRNA levels were also no different between the various groups (Fig. 9B).
FIGURE 9.
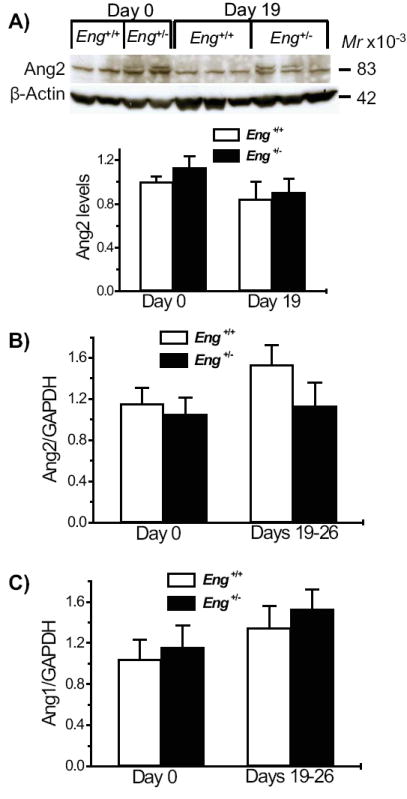
Ang1 and Ang2 levels are similar in the distal colon of Eng+/− and Eng+/+ mice. A) Ang2 levels were measured by Western blot (N = 5-6) under reducing conditions and expressed relative to β-actin and normalized to the level observed in untreated Eng+/+ mice. No significant change was observed between any of the groups. B) Ang 2 and C) Ang1 mRNA levels were estimated by real time PCR. No significant difference was observed between Eng+/− and Eng+/+ mice at days 0 or 19-26. N = 6-7 for Ang2 and N = 7-8 for Ang1 with 2-4 replicates per sample.
Similarly, Ang1 mRNA levels in colonic samples, prior to DSS induction and at day 19 were not significantly different between Eng+/- or Eng+/+ mice. Protein levels could not be measured due to lack of good antibodies to mouse Ang1 (Fig. 9C). Thus Ang1 and Ang2 do not appear to play a significant role in this model of chronic colitis.
DISCUSSION
Our studies revealed that mice expressing a single allele of the Endoglin gene develop persistent, more severe colitis than their control littermates following a single course of DSS treatment. The Eng+/- mice progressed to chronic inflammatory bowel disease, while Eng+/+ mice recovered. The chronic phase of intestinal disease was associated with prominent, pathological angiogenesis, characterized by a high number of vascular protrusions extending from the lamina propria into the serosa and by abnormally dilated vessels in the serosa. We demonstrated that VEGF, but not Ang1 and Ang2, is likely responsible for the pathological angiogenesis associated with chronic bowel inflammation in the Eng+/- mice. Our data also revealed that in the absence of inflammatory stimulus, colonic VEGF levels and vascular permeability were higher in Eng+/- than Eng+/+ mice, which may predispose to subsequent chronic inflammation and sustained pathological angiogenesis.
From the clinical standpoint, colitis was more severe in the Eng+/- mice. More significant losses in body weight and fecal blood, and further shortening of the colon, were seen from day 9 onwards, in the post-acute phase. A lower survival rate was observed in Eng+/- mice compared to Eng+/+ mice from day 12 onwards. Interestingly, the onset of disease was similar in both groups. Progression to severe chronic IBD and a much slower resolution of clinical features of colitis characterized the Eng+/- group. From the histopathological point of view, most animals showed similar extensive leukocyte infiltration and crypt damage at days 5-9 with no significant difference between the Eng+/- and Eng+/+ DSS-treated mice. The angiogenic score, defined by the number of vascular protrusions from mucosa and submucosa into the serosa, was identical between Eng+/- and Eng+/+ mice on days 5 and 9 respectively and correlated well with the inflammation score. These data support the concept that physiological, reparatory angiogenesis is an essential component of an inflammatory response.1
However, the Eng+/- mice, congenic on the C57BL/6 background, progressed steadily to a chronic phase of colitis, demonstrated best between days 19 and 26 after DSS induction, and characterized by even higher inflammation and angiogenic scores than the acute phase. The Eng+/+ C57BL/6 mice did show resolution of intestinal inflammation and a decrease in angiogenesis, at days 19-26. Although a previous study using the same 5-day single course of 3% DSS induction showed that C57BL/6 mice progress from acute to chronic colitis,32 other reports have indicated that repeated cycles of exposure to 2-5% DSS in drinking water for 5 days and interrupted by recovery periods, were needed to induce chronic colitis in these mice.8,33,34
Associated with the chronic phase of colitis in Eng+/- mice, was an expansion of the tissue microvasculature into the serosa, which occurs following increased recruitment of inflammatory cells. Pathological angiogenesis, observed in IBD, is characterized by an irregular vasculature with tortuous vessels, showing increased permeability and inflammatory and thrombogenic potential.35 Increased vessel density, as visualized by Doppler ultrasounds, was documented in the inflamed bowel loops of CD patients.6 Potent angiogenic activity was observed in CD and UC mucosa, further supporting that angiogenesis is an integral component of chronic inflammation in IBD.36 Neovascularization on the serosal surfaces of the chronically inflamed bowel is thought to be the most compelling evidence for angiogenesis playing a role in IBD.37 It is interesting that the Eng+/- mouse model of chronic IBD showed an enlargement of the serosal layer associated with an increased number of invading abnormal vessels, likely providing nutrients and O2 to the growing tissue needed to sustain the persistent inflammation.
VEGF expression was higher in the distal colon of Eng+/- mice compared to Eng+/+ mice, prior to induction of colitis. Mice heterozygous for Endoglin and the functionally related endothelial specific TGF-β superfamily receptor, Alk1, are models for the systemic vascular disorder HHT. Increased plasma levels and tissue staining for VEGF have been reported in HHT patients.19,38 More recently, VEGF was shown to be up-regulated in several tissues including the intestine of Alk1+/- mice.39 Our data of increased VEGF expression in the colon of Eng+/- mice support the view that a sustained increase in tissue VEGF may eventually lead to the vascular malformations associated with HHT. The conditional over-expression of VEGF was shown to lead to the formation of irregular vessels, which may function abnormally.40 VEGF over-expression in the cerebral cortex induced microvessels in both Eng+/- and wild type mice; the difference was that the induced vessels were enlarged and grossly abnormal only in Eng+/- mice.41 Subsequent studies revealed that similarly abnormal microvascular structures were induced by VEGF over-expression in the brain of Alk1+/- mice.42 This suggests that a sustained increase in VEGF in HHT patients may trigger vascular remodeling and eventually induce organ-specific arteriovenous malformations.
Our study suggests no significant involvement of Ang1 and Ang2 in the dysregulated angiogenesis present in Eng+/- mice, prior to DSS induction or in the development of chronic colitis. Although recent data on DSS-induced colitis in Ang2-/- mice suggest that both inflammation-induced hemangiogenesis and lymphangiogenesis are reduced, many clinical signs of intestinal disease, e.g. weight loss, colon length, occult blood, and diarrhea were worse in Ang2-/- than in wild type control mice.43 The exact role of angiopoietins in the development of intestinal inflammation and pathological angiogenesis remains to be elucidated, but does not appear to be significant in the Eng+/- mouse model.
A novel finding in the current study is the significant increase in VP observed in the colon of the Eng+/- mice. There are different types of VP: the low physiological basal VP and the increased VP associated with pathological conditions.44 Basal VP occurs in capillaries while VEGF-A-induced acute vascular hyperpermeability, characteristic of inflammation, takes place in post-capillary venules. Chronic vascular hyperpermeability, which persists in tumor angiogenesis and chronic inflammatory diseases, is mediated by enlarged pericyte-poor thin walled sinusoids derived from pre-existing normal venules after prolonged exposure to VEGF or other angiogenic stimuli.45-47 It is interesting to speculate that the increased VP seen in the Eng+/- mice is due to a persistent increase in VEGF and contributes to the eventual generation of vascular malformations. The further elevation in VEGF, observed in the chronic phase of colitis, may lead to the chronic vascular hyperpermeability associated with pathological angiogenesis and the abnormal vessels observed in the colonic serosa. This would imply that venules rather than capillaries are mediating the increased VP in the Eng+/- mice. Quantitation of the luminal diameter of capillaries in the mucosal plexus of the colonic lamina propria by scanning electron microscopy revealed no change in Eng+/- versus control mice, nor in Eng+/- mice, 9 days after DSS induction (data not shown), suggesting no structural alterations in capillaries under basal conditions and during acute colitis.
TGF-β can regulate VEGF production and there is cross-talk between their signaling pathways.19,20 Endoglin modulates TGF-β responses, particularly in endothelial cells and its insufficiency leads to abnormal angiogenesis. Systemic expression of soluble endoglin, which blocks TGF-β signaling, led to an increase in VP.28 In the current study, we demonstrate that Eng+/- mice show an increase in colonic VP, which is likely mediated by the sustained increase in tissue VEGF, further supporting the cross-talk between TGF-β and VEGF pathways and its critical role in maintaining vascular integrity. Inflammation induced by DSS can further increase VEGF levels and lead to the pathological angiogenesis, which sustains chronic bowel disease in Eng+/- mice. Our data suggest than anti-VEGF therapy may alleviate the changes in VP observed in Eng+/- mice and prevent the progression to chronic inflammation induced by DSS.
Acknowledgments
This work was supported by a grant from the Canadian Institute of Health Research (FRN 6247), and by NIH grant #5 RO1 HL075426. We thank Drs SJ Mentzer for including us as co-applicants on the NIH grant and DJ Ravnic for initiating us to the colitis model. We thank Genentech Inc., San Francisco, CA for providing us with the monoclonal antibody to VEGF-A. We appreciate the helpful discussions with Dr R.S. Kerbel.
References
- 1.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 2.Danese S. Inflammation and the mucosal microcirculation in inflammatory bowel disease: the ebb and flow. Curr Opin Gastroenterol. 2007;23:384–389. doi: 10.1097/MOG.0b013e32810c8de3. [DOI] [PubMed] [Google Scholar]
- 3.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol. 2007;178:6017–6022. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 4.Jackson JR, Seed MP, Kircher CH, et al. The codependence of angiogenesis and chronic inflammation. Faseb J. 1997;11:457–465. [PubMed] [Google Scholar]
- 5.Szekanecz Z, Koch AE. Vascular endothelium and immune responses: implications for inflammation and angiogenesis. Rheum Dis Clin North Am. 2004;30:97–114. doi: 10.1016/S0889-857X(03)00116-9. [DOI] [PubMed] [Google Scholar]
- 6.Spalinger J, Patriquin H, Miron MC, et al. Doppler US in patients with crohn disease: vessel density in the diseased bowel reflects disease activity. Radiology. 2000;217:787–791. doi: 10.1148/radiology.217.3.r00dc19787. [DOI] [PubMed] [Google Scholar]
- 7.Maconi G, Sampietro GM, Russo A, et al. The vascularity of internal fistulae in Crohn’s disease: an in vivo power Doppler ultrasonography assessment. Gut. 2002;50:496–500. doi: 10.1136/gut.50.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chidlow JH, Jr, Langston W, Greer JJ, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. 2006;169:2014–2030. doi: 10.2353/ajpath.2006.051021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136:585–595. e585. doi: 10.1053/j.gastro.2008.09.064. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond) 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 11.Dvorak HF, Detmar M, Claffey KP, et al. Vascular permeability factor/vascular endothelial growth factor: an important mediator of angiogenesis in malignancy and inflammation. Int Arch Allergy Immunol. 1995;107:233–235. doi: 10.1159/000236988. [DOI] [PubMed] [Google Scholar]
- 12.Lee YC. The involvement of VEGF in endothelial permeability: a target for anti-inflammatory therapy. Curr Opin Investig Drugs. 2005;6:1124–1130. [PubMed] [Google Scholar]
- 13.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 14.Koutroubakis IE, Xidakis C, Karmiris K, et al. Potential role of soluble angiopoietin-2 and Tie-2 in patients with inflammatory bowel disease. Eur J Clin Invest. 2006;36:127–132. doi: 10.1111/j.1365-2362.2006.01602.x. [DOI] [PubMed] [Google Scholar]
- 15.Pousa ID, Mate J, Salcedo-Mora X, et al. Role of vascular endothelial growth factor and angiopoietin systems in serum of Crohn’s disease patients. Inflamm Bowel Dis. 2008;14:61–67. doi: 10.1002/ibd.20269. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizaki A, Nakayama T, Naito S, et al. Expression patterns of angiopoietin-1, -2, and tie-2 receptor in ulcerative colitis support involvement of the angiopoietin/tie pathway in the progression of ulcerative colitis. Dig Dis Sci. 2009;54:2094–2099. doi: 10.1007/s10620-008-0593-3. [DOI] [PubMed] [Google Scholar]
- 17.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-ß1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulkarni AB, Karlsson S. Transforming growth factor-β1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]
- 19.Sadick H, Riedel F, Naim R, et al. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica. 2005;90:818–828. [PubMed] [Google Scholar]
- 20.Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res. 2008;102:637–652. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 21.Cheifetz S, Bellón T, Calés C, et al. Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- 22.Toporsian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96:684–692. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 23.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourdeau A, Faughnan ME, McDonald ML, et al. Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am J Pathol. 2001;158:2011–2020. doi: 10.1016/s0002-9440(10)64673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohkawara T, Nishihira J, Takeda H, et al. Geranylgeranylacetone protects mice from dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 2005;40:1049–1057. doi: 10.1080/00365520510023161. [DOI] [PubMed] [Google Scholar]
- 27.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 29.Khazaei M, Nematbakhsh M. Coronary vascular and aortic endothelial permeability during estrogen therapy: a study in DOCA-salt hypertensive ovariectomized rats. Physiol Res. 2004;53:609–614. [PubMed] [Google Scholar]
- 30.Toporsian M, Govindaraju K, Nagi M, et al. Downregulation of endothelial nitric oxide synthase in rat aorta after prolonged hypoxia in vivo. Circ Res. 2000;86:671–675. doi: 10.1161/01.res.86.6.671. [DOI] [PubMed] [Google Scholar]
- 31.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 32.Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 33.Ghia JE, Blennerhassett P, Deng Y, et al. Reactivation of inflammatory bowel disease in a mouse model of depression. Gastroenterology. 2009;136:2280–2288. e2281–2284. doi: 10.1053/j.gastro.2009.02.069. [DOI] [PubMed] [Google Scholar]
- 34.Peng XD, Wu XH, Chen LJ, et al. Inhibition of phosphoinositide 3-kinase ameliorates dextran sodium sulfate-induced colitis in mice. J Pharmacol Exp Ther. 2010;332:46–56. doi: 10.1124/jpet.109.153494. [DOI] [PubMed] [Google Scholar]
- 35.Chidlow JH, Jr, Shukla D, Grisham MB, et al. Pathogenic angiogenesis in IBD and experimental colitis: new ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol. 2007;293:G5–G18. doi: 10.1152/ajpgi.00107.2007. [DOI] [PubMed] [Google Scholar]
- 36.Danese S, Sans M, de la Motte C, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology. 2006;130:2060–2073. doi: 10.1053/j.gastro.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 37.Hatoum OA, Heidemann J, Binion DG. The intestinal microvasculature as a therapeutic target in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:78–97. doi: 10.1196/annals.1326.003. [DOI] [PubMed] [Google Scholar]
- 38.Sadick H, Naim R, Gossler U, et al. Angiogenesis in hereditary hemorrhagic telangiectasia: VEGF165 plasma concentration in correlation to the VEGF expression and microvessel density. Int J Mol Med. 2005;15:15–19. [PubMed] [Google Scholar]
- 39.Shao ES, Lin L, Yao Y, et al. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood. 2009;114:2197–2206. doi: 10.1182/blood-2009-01-199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dor Y, Djonov V, Abramovitch R, et al. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu B, Wu YQ, Huey M, et al. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab. 2004;24:237–244. doi: 10.1097/01.WCB.0000107730.66603.51. [DOI] [PubMed] [Google Scholar]
- 42.Hao Q, Su H, Marchuk DA, et al. Increased tissue perfusion promotes capillary dysplasia in the ALK1-deficient mouse brain following VEGF stimulation. Am J Physiol Heart Circ Physiol. 2008;295:H2250–2256. doi: 10.1152/ajpheart.00083.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganta VC, Cromer W, Mills GL, et al. Angiopoietin-2 in experimental colitis. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.21150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy JA, Benjamin L, Zeng H, et al. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dvorak HF, Nagy JA, Feng D, et al. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 46.Pettersson A, Nagy JA, Brown LF, et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 47.Feng D, Nagy JA, Dvorak AM, et al. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res. 2000;59:24–37. doi: 10.1006/mvre.1999.2207. [DOI] [PubMed] [Google Scholar]