

Abstract
Background
Acute severe ulcerative colitis (UC) remains a significant clinical challenge and the ability to predict, at an early stage, those individuals at risk of colectomy for medically refractory UC (MR-UC) would be a major clinical advance. The aim of this study was to use a genome-wide association study (GWAS) in a well-characterized cohort of UC patients to identify genetic variation that contributes to MR-UC.
Methods
A GWAS comparing 324 MR-UC patients with 537 Non-MR-UC patients was analyzed using logistic regression and Cox proportional hazards methods. In addition, the MR-UC patients were compared with 2601 healthy controls.
Results
MR-UC was associated with more extensive disease (p= 2.7×10−6) and a positive family history of UC (p= 0.004). A risk score based on the combination of 46 SNPs associated with MR-UC explained 48% of the variance for colectomy risk in our cohort. Risk scores divided into quarters showed the risk of colectomy to be 0%, 17%, 74% and 100% in the four groups. Comparison of the MR-UC subjects with healthy controls confirmed the contribution of the major histocompatibility complex to severe UC (peak association: rs17207986, p= 1.4×10−16) and provided genome-wide suggestive association at the TNFSF15 (TL1A) locus (peak association: rs11554257, p= 1.4×10−6).
Conclusions
A SNP-based risk scoring system, identified here by GWAS analyses, may provide a useful adjunct to clinical parameters for predicting natural history in UC. Furthermore, discovery of genetic processes underlying disease severity may help to identify pathways for novel therapeutic intervention in severe UC.
Keywords: Ulcerative colitis, Natural History, Genetics
Introduction
Genome-wide association studies (GWAS) have advanced our knowledge of the genetic contribution to ulcerative colitis (UC) and led to the identification of approximately 20 UC-associated loci, including the major histocompatibility complex (MHC) 1–6. These advances in UC, in addition to those made in CD, have significantly increased our understanding of the underlying pathogenic processes that lead to chronic mucosal inflammation. Yet despite the identification of approximately 50 IBD susceptibility loci, there has been minimal clinical benefit for IBD patients or clinicians 4, 7.
Patients with UC demonstrate a degree of heterogeneity as age of onset, disease extent, natural history, response to medical therapies, and need for surgery vary between individuals. Medically refractory ulcerative colitis (MR-UC) requiring colectomy remains a significant challenge in the management of IBD. In one five-year study, more than 12% of ~1100 UC patients developed toxic, fulminant or severe colitis 8; in a population-based study in Norway, 7.5% of approximately 450 UC patients followed up for five years required colectomy 9; and in another study from Copenhagen, more than 30% of UC patients required colectomy 18 years after diagnosis 10. While a number of prognostic models have been developed for predicting outcomes in acutely severe UC, it would be a significant advance if patients at high risk for severe disease could be identified earlier in their course of disease, as they may benefit from an earlier introduction of more intensive therapy and monitoring. Previous studies have shown that severe UC is more prevalent in non-smokers and ex-smokers, in those with an extensive distribution of disease 11, 12, and may also be more common in Caucasians than Hispanics and Asians 13, 14. However, none of these parameters alone demonstrate adequate clinical utility.
A number of studies have examined the role of genetic variation in the development of extensive and severe UC and have implicated loci including the MHC and the multidrug resistance gene 1 (MDR1/ABCB1) 15–23. These data support the hypothesis that genetic variation contributes not only to UC susceptibility, but also to clinical phenotype and natural history. Furthermore, recent advances in IBD genetics have confirmed that genetically complex conditions such as IBD are caused by multiple genetic variants. A number of studies combining information from multiple loci have shown some utility in other complex conditions such as diabetes 24–26. Crohn’s disease investigators, including our own group, have started utilizing these approaches for predicting both natural history in and response to anti-TNF therapy in CD 27, 28. In this study, we aimed to uncover genetic associations with MR-UC both to assess their utility in identifying patients at an increased risk of colectomy and to identify potential novel therapeutic targets for the treatment of severe UC.
Materials and Methods
Subjects
UC Cases
Ulcerative Colitis (UC) subjects (n= 929) were recruited at Cedars Sinai-Medical Center Inflammatory Bowel Disease Center following informed consent after approval by the Institutional Review Board. UC diagnosis was based on standard criteria29. UC subjects requiring colectomy for severe disease refractory to medical therapies were classified as medically refractory UC (MR-UC). Approximately 66% of the MR-UC patients had been treated with cyclosporine and 24% with biologic therapies prior to colectomy. Subjects requiring colectomy where the indication was for treatment of cancer/dysplasia, in addition to subjects not requiring colectomy, were classified as Non-MR-UC. Subjects who required colectomy for MR-UC and were subsequently found to have evidence of dysplasia or carcinoma in the resected colon were classified as MR-UC (n= 3). For the MR-UC cohort, time from diagnosis to date of colectomy was collected; time from diagnosis to last follow-up visit was obtained for the Non-MR-UC cohort. Samples which did not genotype successfully (n= 16), exhibited gender mismatch (n= 9) or cryptic relatedness (n= 13), or were considered outliers by principal components analysis (n= 30; see below) were excluded. Following these measures, 861 UC subjects (MR-UC n= 324; Non-MR-UC n= 537) were included in the analyses.
Non-IBD Controls
Controls were obtained from the Cardiovascular Health Study (CHS), a population-based cohort study of risk factors for cardiovascular disease and stroke in adults 65 years of age or older, recruited at four field centers 30, 31. 5,201 predominantly Caucasian individuals were recruited in 1989–1990 from random samples of Medicare eligibility lists, followed by an additional 687 African-Americans recruited in 1992–1993 (total n= 5,888). CHS was approved by the Institutional Review Board at each recruitment site, and subjects provided informed consent for the use of their genetic information. A total of 2,601 Caucasian non-IBD control subjects who underwent GWAS were included in these analyses. African-American CHS participants were excluded from analysis due to insufficient number of ethnically-matched cases.
Genotyping
All genotyping was performed at the Medical Genetics Institute at Cedars-Sinai Medical Center using Infinium technology (Illumina, San Diego, CA) 32, 33. UC cases were genotyped with either the HumanCNV370-Quad or Human610-Quad platform; controls were genotyped with the HumanCNV370-Duo platform. Identity-by-descent was used to exclude related individuals (Pi-hat scores >0.5; PLINK) 34. Average genotyping rate among cases and controls retained in the analysis was >99.8% and >99.2%, respectively. Single nucleotide polymorphisms (SNPs) were excluded based on: test of Hardy-Weinberg Equilibrium p <10−3; SNP failure rate >10%; MAF <3%; SNPs not found in dbSNP Build 129. 313,720 SNPs passed quality control measures and were common in all data sets.
Statistical Analyses
Population Stratification
Principal components analysis (Eigenstrat as implemented in Helix Tree) (Golden Helix, Bozeman, MT) was conducted to examine population stratification 35. Extreme outliers, defined as subjects more than two standard deviations away from the distribution of the rest of the samples for any component, were removed. All African-American participants identified by principal components analysis were excluded from these analyses. Genetic heterogeneity following correction for population sub-structure was low, with estimated genomic inflation factors (λGC) of 1.04 and 1.06 for MR-UC vs. Non-MR-UC, and MR-UC cases vs. Non-IBD controls analyses, respectively 36.
MR-UC vs. Non-MR-UC: Survival Analysis and Risk Modeling
Single marker association analysis of MR-UC vs. Non-MR-UC (analysis-I) was performed using a logistic regression model correcting for population stratification using 20 principal components as covariates (PLINK v1.06) 34. Association between medically refractory disease (MR-UC) and the top 100 SNPs together (as determined by the lowest corrected p-values) from analysis-I were tested using a stepwise logistic regression model. SNPs were further analyzed by Cox proportional hazards regression utilizing time-to information, as described above for UC cases (using the step and glm, and coxph functions, respectively, in R v2.9.0) 37. 37 SNPs identified with logistic regression p <0.05 and Cox proportional hazards p <0.1 were retained in the risk model. The 100 SNPs (p <3×10−4) evaluated from analysis-I are listed in Supplementary Table S1. A genome-wide Cox proportional hazards regression analysis (analysis-II) was then performed on a subset of our UC cohort (MR-UC subjects with colectomy <60 months, n= 187; Non-MR-UC followed up >60 months, n= 328) correcting for population stratification using two principal components as covariates (PLINK). The top 65 SNPs (8 of which overlap with the 100 SNPs from analysis-I above) were tested together (using coxph function in R). The 65 SNPs (p <1×10−4) from analysis-II are listed in Supplementary Table S2. From these 65 SNPs, 9 SNPs were identified (p <3×10−4) and combined with the 37 SNPs from analysis-I to identify a final risk model consisting of 46 SNPs (see Figure 1 for schematic). A genetic risk score was calculated from the total number of risk alleles (0, 1, or 2) across all 46 risk SNPs (theoretical range: 0–92). Risk score (observed range: 28–60) was divided into quarters: scores 28–38 (risk-A); scores 39–45 (risk-B); scores 46–52 (risk-C); and scores 53–60 (risk-D). Receiver operating characteristic (ROC) curve and area under the ROC curve (AUC) were calculated using R software v2.9.0, including packages survival and survivalROC 37–39. Sensitivity and specificity curves, positive and negative predictive values, positive (sensitivity/1−specificity) and negative likelihood ratio (1−sensitivity/specificity) were all calculated using the R package ROCR 40. 1000-fold replication of 10-fold cross-validation was implemented to validate the fitted logistic regression model. Mean sensitivity and specificity were then re-calculated using the 1000 replicated samples. Bootstrap method with 1000-fold replication was utilized for estimating variability of hazard ratio estimated from the Cox regression model 41, 42. The hazard ratio in survival analysis is the effect of an explanatory variable on the hazard or risk of an event.
Figure 1.
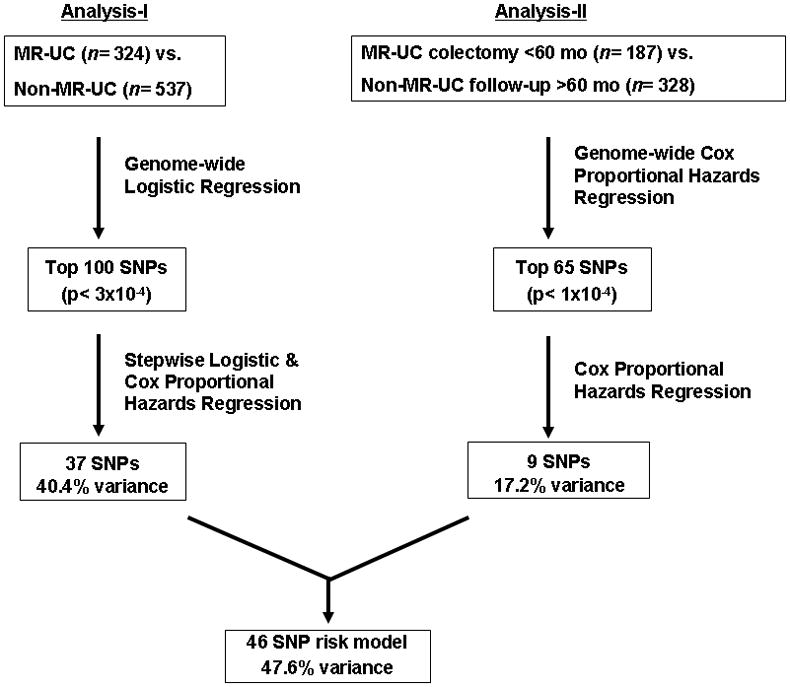
Schematic describing MR-UC vs. Non-MR-UC survival analysis and risk modeling
MR-UC vs. Non-IBD Controls: Regression Analysis
Single marker analysis of genome-wide data for MR-UC cases vs. Non-IBD Caucasian controls from CHS (analysis-III) was performed as above, using logistic regression correcting for 20 principal components (PLINK).
Results
UC subject demographics
Complete temporal data was available on 861 UC subjects (MR-UC n= 324; Non-MR-UC n= 537). The demographic data of our cohort is summarized in Table 1. We observed no differences in gender, median age of onset of disease, and smoking status between our medically refractory and Non-MR-UC subjects. There was a significant difference in our median disease duration (p= 7.4×10−9), with the time from diagnosis to last follow-up in the Non-MR-UC cohort nearly double the time from diagnosis to colectomy in our MR-UC subjects. Additionally, there was a significantly higher incidence of disease that extended proximal to the splenic flexure (p= 2.7×10−6) in the MR-UC group when compared to Non-MR-UC, consistent with previously published data 43. We identified a novel association between a family history (first or second degree relative) of UC and the development of MR-UC (p= 0.004).
Table 1.
UC subject demographics. (IQR=interquartile range; P-values calculated using logistic regression.)
| FACTORS | Non-MR-UC (n=537) | MR-UC (n=324) | P-value |
|---|---|---|---|
| Sex (F%) | 47% | 47% | 0.89 |
| Median Age of Onset - yrs (IQR) | 26 (17–38) | 27 (19–37) | 0.93 |
| Smoking (%) | 8% | 6% | 0.24 |
| Median Disease Duration - months (IQR) | 95 (42–198) | 48 (24–120) | 7.4×10-9 |
| Extensive Disease (%) | 64% | 80% | 2.7×10-6 |
| Family History of IBD (%) | 21% | 32% | 0.0009 |
| Family History of UC (%) | 15% | 24% | 0.004 |
Forty-six SNP risk model is associated with MR-UC and predicts earlier progression to colectomy
We performed a GWAS on 324 MR-UC and 537 Non-MR-UC subjects. Results of this analysis (analysis-I) are given in Table S1 and discussed below. Following identification of single markers associated with MR-UC, we proceeded to a multivariate approach. Beginning with the top 100 results from analysis-I (p <3×10−4), we performed a stepwise logistic regression and identified 64 SNPs (all had p <0.05) that together were associated with medically refractory disease (MR-UC) and were carried forward to survival analysis. Of these 64 SNPs, 37 SNPs remained (Cox proportional hazards regression p <0.1; OR 1.2–1.8), which explained 40% of the variance for MR-UC (Table 2, see non-bolded SNPs). In order to elucidate the maximum discrimination, i.e. greatest percentage of the variance, we further performed a genome-wide Cox proportional hazards regression analysis (analysis-II) on a subset of our UC cohort (see Methods) to identify SNPs involved in earlier progression to colectomy. Testing together the top 65 SNPs from this analysis (Table S2, p <1×10−4), we identified nine SNPs with Cox proportional hazards p <3×10−4 (individual OR ranged from 1.4–1.6), explaining 17% of the variance (Table 2, see bolded SNPs). Beginning with our previously identified 37 risk SNP model, these 9 SNPs were added sequentially to the model. This analysis resulted in the final risk model consisting of 46 SNPs (OR for MR-UC for each individual SNP ranged from 1.2–1.9), which explained 48% of the variance for colectomy in our MR-UC cohort (Table 2).
Table 2.
46 Risk SNPs for MR-UC
| Chr* | SNP | Position* | Risk_allele | Loci ** |
|---|---|---|---|---|
| 1 | rs746503 | 54842574 | A | ACOT11 | FAM151A | C1orf175 | 645442 | |
| 1 | rs2275612 | 95140004 | A | CNN3 | SLC44A3 | 646896 | 729970 | |
| 1 | rs7550055 | 157045388 | C | MNDA | OR6N2 | OR2AQ1P | OR10AA1P | OR6K4P | OR6N1 | OR6K3 | OR6K5P | 646377 | |
| 1 | rs7367845 | 224512151 | A | ACBD3 | MIXL1 | LIN9 | 100128832 | |
| 2 | rs1448901 | 206961885 | G | ADAM23 | 100132849 | |
| 2 | rs4487082 | 229432205 | A | 2q36.3 |
| 3 | rs900569 | 41834977 | G | ULK4 | |
| 3 | rs924022 | 65824936 | A | MAGI1 | |
| 3 | rs9843732 | 135505746 | G | RYK | |
| 4 | rs2286461 | 15572771 | G | PROM1 | FGFBP1 | FGFBP2 | 100130067 | |
| 4 | rs12650313 | 41401850 | A | LIMCH1 | 100128654 | |
| 4 | rs1399403 | 108639264 | A | 4q25 |
| 4 | rs7675371 | 116049368 | G | NDST4 | |
| 5 | rs3846599 | 10308821 | A | MARCH6 | CCT5 | FAM173B | MIR378 | |
| 5 | rs6596684 | 105972832 | G | 345571 | |
| 6 | rs1536242 | 6876009 | G | 6p25.1 |
| 6 | rs17207986 | 32187545 | G | ATF6B | RNF5 | PPT2 | EGFL8 | 653033 | |
| 6 | rs777649 | 68925053 | A | 642902 | |
| 7 | rs11764116 | 18766938 | A | HDAC9 | |
| 7 | rs4722456 | 25338225 | G | 100131016 | |
| 7 | rs929351 | 81695829 | A | CACNA2D1 | |
| 8 | rs2980654 | 6480608 | G | ANGPT2 | AGPAT5 | MCPH1 | 100131112 | 100132301 | |
| 8 | rs6994721 | 76220268 | A | 8q21.11 |
| 8 | rs4734754 | 105347978 | A | RIMS2 | TM7SF4 | |
| 9 | rs7861972 | 6759692 | A | JMJD2C | SNRPEL1 | |
| 9 | rs3118292 | 25133480 | G | 9p21.3 |
| 9 | rs10817934 | 118589872 | G | ASTN2 | |
| 11 | rs2403456 | 11134390 | A | 11p15.3 |
| 11 | rs1461898 | 37546808 | G | 100132895 | 100132631 | |
| 11 | rs6591765 | 62674829 | G | SLC22A24 | SLC22A25 | SLC22A10 | |
| 12 | rs887357 | 3344906 | A | 643119 | 728230 | 100128253 | |
| 12 | rs526058 | 24326688 | G | 12p12.1 |
| 13 | rs7319358 | 78448935 | A | 13q31.1 |
| 14 | rs1956388 | 28202628 | G | 14q12 |
| 14 | rs11156667 | 30906111 | G | GPR33 | HEATR5A | NUBPL | C14orf126 | 728852 | |
| 14 | rs10133064 | 85844148 | C | 14q31.3 |
| 14 | rs8020281 | 94436179 | A | 14q32.13 |
| 15 | rs965353 | 57847498 | G | BNIP2 | 100130107 | GTF2A2 | |
| 16 | rs305087 | 84539747 | A | 100131952 | |
| 17 | rs759258 | 52483547 | A | AKAP1 | |
| 19 | rs2967682 | 8644532 | C | MYO1F | ADAMTS10 | OR2Z1 | 390880 | |
| 19 | rs2293683 | 12900284 | G | CALR | DNASE2 | GCDH | FARSA | NFIX | RAD23A | PRDX2 | KLF1 | RTBDN | GADD45GIP1 | DAND5 | SYCE2 | |
| 20 | rs6034134 | 15182479 | C | MACROD2 | |
| 20 | rs10485594 | 19772393 | A | RIN2 | 644298 | |
| 20 | rs6059104 | 31185354 | G | PLUNC | C20orf71 | C20orf70 | C20orf186 | 317716 | 391242 | |
| 21 | rs2831462 | 28370367 | A | 21q21.3 |
Defined using dbSNP Build 129.
Loci in potential LD with risk SNP (r2>0.7). Loci include genes that overlap each SNP within 100kb of 5′ end and 10kb of 3′ end of each gene. Numbers refer to dbGene (NCBI). SNPs identified from analysis-II are in bold.
We calculated a genetic risk score from the total number of risk alleles across all 46 risk SNPs (theoretical range: 0–92). The observed risk score ranged from 28–60, and was significantly associated with MR-UC (logistic regression and Cox proportional hazards p-values <10−16). An ROC curve using this risk score gave an AUC of 0.91. The sensitivity of the fitted model for MR-UC was 0.793, with a specificity of 0.858. Using 1000 replicates of the 10-fold cross-validation data, we obtained a mean sensitivity of 0.789 (SD= 0.0067) and mean specificity of 0.859 (SD= 0.002; Table 3). This indicates that the fitted model was robust and only ~0.4% over-fitting was observed. The hazard ratio was estimated to be 1.313 from the Cox regression model. 1000 replicates of bootstrapped samples gave an estimated hazard ratio of 1.314 (SD= 0.017; Table 3).
Table 3.
Sensitivity, Specificity, and Hazard Ratio of original data and following 10-fold Cross-Validation and Bootstrapping.
| Sensitivity/Specificity (cut-off=0.5) | |
|---|---|
| Original Data with logistic regression | |
| Sensitivity | 0.793 |
| Specificity | 0.858 |
| 1000 times of 10 fold Cross-Validation data sets with logistic regression | |||||
|---|---|---|---|---|---|
| Variable | N | Mean | Std Dev | Minimum | Maximum |
| Sensitivity | 1000 | 0.789 | 0.0067 | 0.758 | 0.793 |
| Specificity | 1000 | 0.859 | 0.0021 | 0.858 | 0.870 |
| Hazard Ratio |
|---|
| Original Data |
| 1.313 |
| 1000 fold Bootstrapping | ||||
|---|---|---|---|---|
| N | Mean | Std Dev | Minimum | Maximum |
| 1000 | 1.314 | 0.017 | 1.269 | 1.372 |
Based on the genetic risk scores, we grouped our UC cohort into four risk categories (see Methods); less than 1% of cases in the lowest risk category (risk-A) were MR-UC and the percentage of MR-UC increased to ~17%, ~74% and 100% in risk-B, -C and -D groups, respectively (Figure 2A; χ2 test for trend p <2.2×10−16). The median time to colectomy for risk-C and -D categories was 72 months and 23 months, respectively (given the low incidence of MR-UC in groups -A and -B, it was not possible to calculate the median time to colectomy for these two groups). Progression to colectomy within 2 and 5 years of diagnosis may be more clinically relevant and while no individuals in the risk-A category had undergone colectomy at either 2 or 5 years after diagnosis, the respective incidence of MR-UC at 2 years for risk groups -B, -C and -D was 3.1%, 19.1%, and 62%, respectively, and at 5 years was 8.3%, 50%, and 80%, respectively (Figure 2B). At five years from diagnosis, either the total risk score (AUC 0.86) or the risk category (AUC 0.82) are able to predict patients that will require surgery. The operating characteristics of the risk score system are shown in Supplementary Figure 1. A score of 44 (Figure S1-C) and 47 (Figure S1-D) can be used to generate a test with a sensitivity (to exclude a diagnosis of colectomy) and specificity (to include a diagnosis) of over 90%, respectively.
Figure 2.
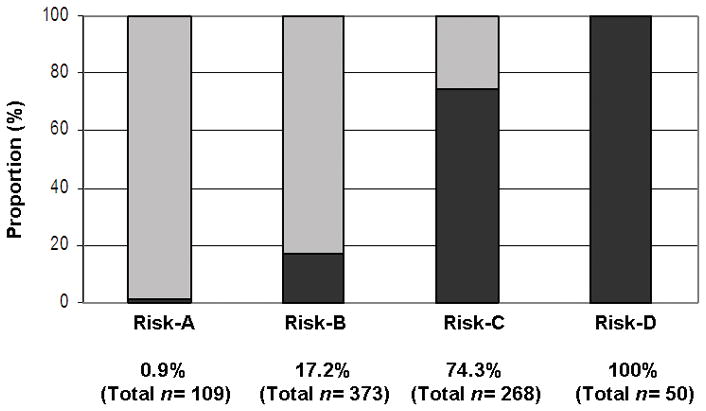
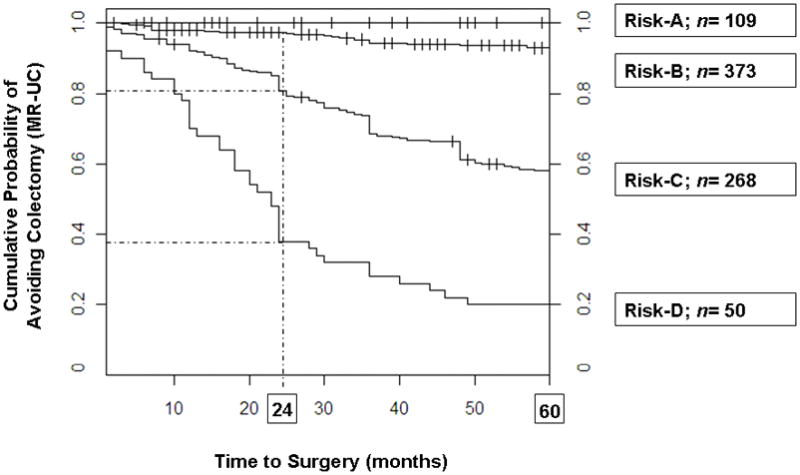
A) Higher risk score categories are associated with MR-UC (χ 2 test for trend p <2.2×10−16). Risk score (observed range: 28–60) was divided into quarters: scores 28–38 (risk-A); scores 39–45 (risk-B); scores 46–52 (risk-C); and scores 53–60 (risk-D). MR-UC, dark grey; Non-MR-UC, light gray. Percentage of MR-UC is noted, along with the total number of UC subjects in each risk category. B) Higher risk score categories are associated with an earlier progression to colectomy at 24 and 60 months. Risk score was divided into quarters (see A above). At 24 months, risk of colectomy was 3.1%, 19.1% and 62% for risk-B, -C, and -D, respectively. Risk of colectomy at 60 months increased to 8.3%, 48.4%, 84% for risk-B, -C, and -D, respectively. Total number of UC subjects in each risk category is given.
Loci corresponding to the 46 SNPs in our risk model include several compelling candidate genes for UC severity and suggest potential biological pathways for further avenues of study (shown Table 2 and discussed later). As each risk SNP contributes only modestly to the overall risk of MR-UC (OR 1.2–1.9), this work supports the paradigm that a group of SNPs, identified by GWAS and combined together may account for a large proportion of the genetic contribution to a complex phenotype (48% of the variance for risk in this study) to provide a risk score with clinical utility.
MHC region and TL1A (TNFSF15) contribute to UC severity
Association analyses between 324 UC subjects with MR-UC and 2,601 population-matched controls confirmed a major contribution of the major histocompatibility (MHC) on chromosome 6p to the development of severe UC (analysis-III, Figure 3). Ten SNPs in MHC reached a priori defined level of genome-wide significance (p≤5×10−7; 82 SNPs with p <1×10−3), with peak association at rs17207986 (p= 1.4×10−16; Figure 4A and Supplementary Table S3). Three SNPs on chromosome 9q32, a locus which contains the known IBD susceptibility gene TNFSF15 (TL1A), achieved genome-wide suggestive significance (p <5×10−5), with the most significant association seen at rs11554257 (p= 1.4×10−6; Figure 4B). In addition, we observed association with several known and putative UC loci, including interleukin (IL)-10 (1q32.1), IL-12B (5q33.3), 12q15 (IFNG/IL-26), ZFP90 (16q22.1) and GSDML/ORMDL3 (17q12) when comparing MR-UC and controls 1, 3, 4, 44, 45. An association with ZFP90 was also observed in our analysis of MR-UC and Non-MR-UC. Furthermore, we confirmed association with a newly identified UC locus, KIF1A (2q37.3), in our association analysis of MR-UC and Non-MR-UC 44 (Table 4).
Figure 3.
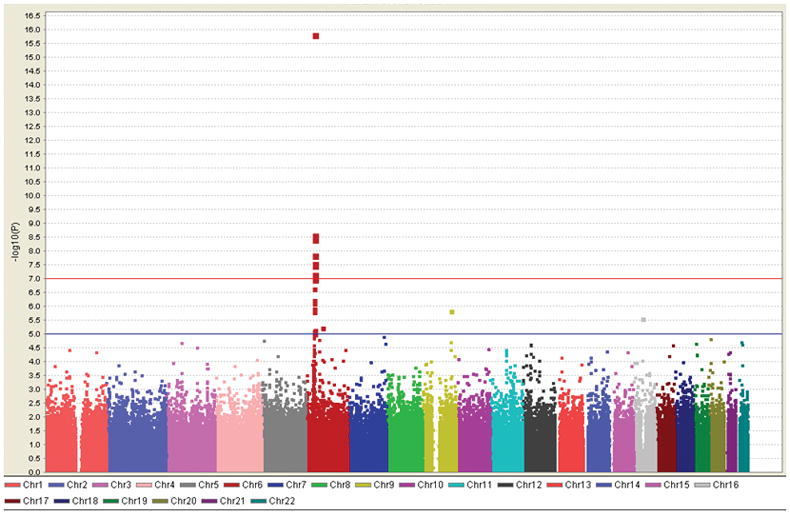
Manhattan plot depicting MR-UC vs. Control analysis, which confirms a significant contribution of the MHC region (chromosome 6) to UC severity, with suggestive significance observed for TNFSF15 (TL1A) region on chromosome 9. Red line denotes 10−7, a level of genome-wide significance; blue line denotes suggestive significance at 10−5. (Generated using Haploview 71.)
Figure 4.
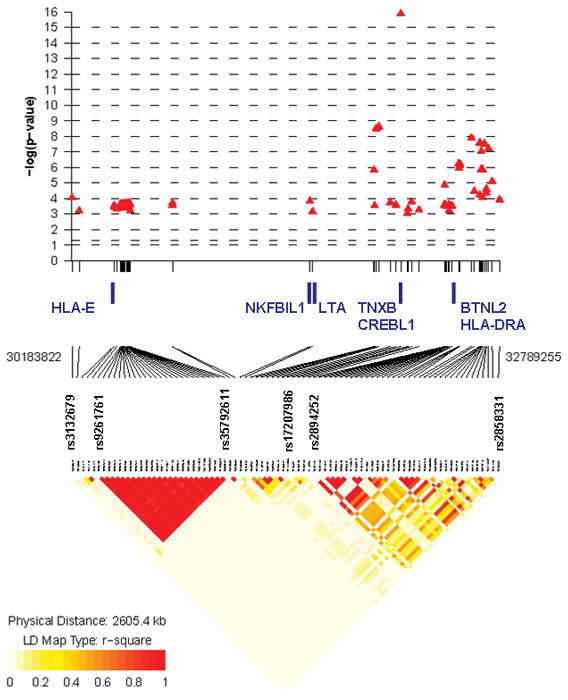
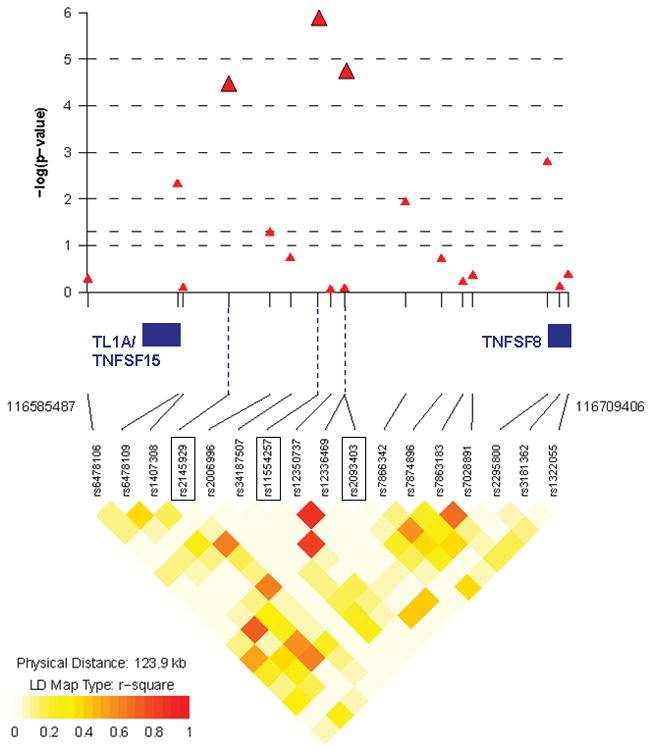
A) P-values and LD plot of 82 SNPs (p <1×10−3) on chromosome 6p of MHC region from MR-UC vs. healthy controls (analysis-III). Ten SNPs reached genome-wide significance (p ≤5×10−7), with an association peak at rs17207986 (p= 1.4×10−16). Leading and trailing SNPs, and SNPs bordering haplotype blocks are further identified (see Table S3 for complete list of SNPs). B) P-values and LD plot of 17 SNPs (p <1×10−3) within the TL1A/TNFSF15 and TNFSF8 regions on chromosome 9q from MR-UC vs. healthy controls (analysis-III). Three SNPs reached genome-wide suggestive significance (p <5×10−5): rs2145929 (p= 3.7×10−5), rs2093403 (p= 1.9×10−5), with an association peak at rs11554257 (p= 1.4×10−6). SNPs are represented as red triangles. LD heat map indicates level of linkage disequilibrium (r-squared) between SNPs. Chromosomal position of leading and trailing SNPs, and genes of interest located within this region are noted. (Generated with R package snp.plotter; http://cbdb.nimh.nih.gov/~kristin/snp.plotter.html)
Table 4.
Association between MR-UC and known UC loci
| Chr* | SNP | Position* | Minor Allele | P-value | Odds Ratio | Loci of Interest | Reference(s) |
|---|---|---|---|---|---|---|---|
| 1 | rs3024505 | 205006527 | A | 2.21E-03 | 1.43 | IL-10 | 3, 44 |
| 2 | rs2288750 | 241371118 | A | 2.07E-03 | 1.51 | KIF1A | 44 |
| 5 | rs7709212 | 158696755 | G | 1.29E-03 | 1.36 | IL-12B | 1 |
| 6 | rs28772724 | 32617335 | A | 1.34E-08 | 0.48 | MHC | 2, 3, 4, 6 |
| 9 | rs11554257** | 116644891 | G | 1.39E-06 | 1.82 | TNFSF15 | 64 |
| 12 | rs1558744 | 66790859 | A | 9.03E-05 | 1.45 | IFNG | IL-26 | 4 |
| 16 | rs1182968** | 67131446 | G | 5.66E-04 | 0.66 | ZFP90 | 45 |
| 17 | rs2305480 | 35315722 | A | 1.43E-03 | 1.35 | GSDML | ORMDL3 | 44 |
Association results observed in MR-UC vs. Non-IBD controls (analysis-III), with the exception of rs2288750, which was observed in MR-UC vs. Non-MR-UC analysis-I only (in bold).
SNP found to be associated in both analysis-I and –III.
Defined using dbSNP Build 129.
Discussion
Utilizing a GWAS approach of a well-characterized UC cohort and a large healthy control group, we confirmed the contribution of the MHC to severe UC at a genome-wide level of significance and our data suggest that there is more than one severity associated ‘signal’ from this locus. We also implicated TNFSF15 (TL1A) in UC severity, with potential therapeutic implications 46. We confirmed that extensive disease and identified that a family history of UC are associated with the need for surgery (Table 1), justifying our hypothesis that genetic variation contributes to the natural history of UC. The 46 SNP model discriminates patients at risk of MR-UC and explains approximately 50% of the genetic contribution to the risk of surgery in our cohort. Higher risk score categories had an elevated percentage of MR-UC subjects (p <2.2×10−16; Figure 2A) and predicted earlier colectomy (Figure 2B).
The predictive power of diagnostic tests can be evaluated by the area under the curve (AUC), an ROC summary index, which evaluates the probability that one’s test correctly identifies a diseased subject from a pair of affected and unaffected individuals. A perfect test has an AUC of 1.0, while random chance gives an AUC of 0.5 39, 47. Screening programs attempting to identify high-risk groups generally have an AUC of ~0.80 48. The genetic risk score reported here yielded an AUC of 0.91. Furthermore, we calculated operating characteristics for our model (see Methods; Table 3 and Figure S1) and ‘scores’ of 44 and 47 (out of a possible score of 60; Figure S1-C and D) demonstrate a sensitivity and specificity of over 90%, respectively. The fitted model was robust, given the comparable mean sensitivity and specificity following cross-validation (Table 3). In addition, likelihood ratios can be used with differing pre-test probabilities to calculate relevant post-test probabilities and are therefore much more generalizable. The Cochrane collaboration has suggested that positive likelihood ratios of greater than 10 and negative likelihood ratios of less than 0.1 are likely to make a significant impact on health care (reviewed in 49). In our model, these ratios are met with a risk score of 47 and 43, respectively (Figure S1-A and B).
The utility of a model with this ability to discriminate can be demonstrated with the following example: a newly diagnosed patient with UC is estimated to have a pre-test probability of colectomy for MR-UC of approximately 20% (based on epidemiological and clinical data) and has a genetic risk score of 47 (positive likelihood ratio of approximately 10); utilizing Bayesian principles, this equates to a post-test probability of colectomy of approximately 75%. If patients at high risk for colectomy could be identified early in their course of disease, then this could have significant consequences for clinicians who might consider earlier introduction of more potent medication and more intense monitoring for high risk patients. Furthermore, appropriate counseling regarding the risk of colectomy (early introduction to stoma therapists, etc.), the importance of medication compliance, and the need for protecting the anal sphincter in obstetric management may also be appropriate for these patients.
Recent advances in IBD genetics have identified novel pathways involved in disease pathogenesis, and similarly genes associated with more severe disease may highlight new therapeutic targets. A number of interesting genes are highlighted in our analysis comparing MR-UC and Non-MR-UC (see Table S1 and Table 2). Bicaudal-D1 (BICD1), implicated in transport from the Golgi apparatus to the endoplasmic reticulum, has been shown to localize to Chlamydia trachomatis inclusions, suggesting a potential role for BICD1 in the bacterial-host interface 50, 51. Recently, our own group and others have implicated genes encoding proteins involved in epithelial barrier integrity, an important component of innate immunity, in UC pathogenesis 45, 53–55. The identification of MAGI1, a scaffolding protein at cell-cell junctions that localizes at the tight junctions of intestinal epithelial cells, provides further evidence for this phenomenon 52. Retinoid-related orphan receptors (ROR) are a family of nuclear hormone receptors that have an established role in autoimmune disease 56 and in particular ROR-α (associated with MR-UC) plays a critical role in cellular stress response and is highly expressed in Th17 cells, and upregulates IL-17 and IL-17F expression 57. Furthermore, the association between FGFBP2 (KSP37) and MR-UC supports a role for cytotoxic lymphocyte-mediated immunity 58 and interestingly, increased FGFBP2 expression is found in asthma 59.
We identified additional genes of interest when comparing the ‘extreme phenotypes’ of MR-UC and healthy individuals including variants in the MHC, TNFSF15 (TL1A), PLCB1 and CTSF. The MHC relationship with severe UC confirmed earlier associations 2–5, 15–20, 60, 61, with genome-wide levels of significance for ten SNPs (p≤5×10−7; Figure 3 and Table S3) and the suggestion of independent signals encompassing both the HLA-E and BTNL2/HLA-DRA regions (Figure 4A). TNFSF15 (TL1A) is also implicated in severe UC (17 SNPs associated with MR-UC; p <1×10−3), with three SNPs showing suggestive evidence of genome-wide significance (p <5×10−5; Figure 4B). TNFSF15 variants are associated with both CD and UC in a broad spectrum of populations 62–65 and increased TNFSF15 mRNA and protein expression have been demonstrated in inflamed mucosa of the colon and small bowel in CD patients 66, 67. Increased TNFSF15 expression is also correlated with severity of ileal and colonic inflammation in the mouse, supporting our finding of an association with severe disease 46, 66. Administration of neutralizing TNFSF15 antibodies prevent and treat established chronic intestinal inflammation, by suppressing the production of interferon-gamma and IL-17/IL-6 46. Taken together, these data suggest that therapies directed against TNFSF15 may prove useful in the setting of acute severe UC.
Also implicated in severe UC is phospholipase C-β1 (PLCB1; rs6039206, p= 1.5×10−5), an intracellular transductor of extracellular signals, including IL-1β and TNF-α in rheumatoid arthritis 68. Furthermore, PLCB1 has been implicated as a potential asthma modifier gene 69. We also observed a cluster of SNPs at the CTSF locus (association peak at rs540874, p= 4.5×10−5). CTSF (cathespin-F) represents a major component of the lysosomal proteolytic system, a critical determinant of the timing and location of MHC class II peptide loading in macrophages, further implicating the critical role that the MHC plays in disease severity 70.
We have confirmed the association with the MHC and disease severity in UC and our data suggest that there may be more than one ‘signal’ from this locus. Furthermore, we have also implicated a realistic therapeutic target and known IBD locus, TNFSF15 (TL1A), suggesting that interference with this pathway is a worthy avenue for study in severe UC. In addition, we have demonstrated the utility of a model based on GWAS data for predicting the need for surgery in UC, although it is important to acknowledge that these findings clearly require replication in independent cohorts. However, these data suggest that even though the individual effect of any severity associated variant is likely to be small, cumulatively they may provide adequate discriminatory power for clinical use. These findings, if reproduced, would allow a more tailored approach to the management of UC patients, potentially provide a ‘blueprint’ for the utilization of large-scale genomic data in clinical practice, as well as potentially identifying additional targets for early therapeutic intervention in more aggressive UC.
Supplementary Material
The clinical utility of the MR-UC risk score is demonstrated by positive (A) and negative (B) likelihood ratios; sensitivity (C) and specificity (D); and positive (E) and negative (F) predictive values. A score of 44 (C) and 47 (D) can be used to generate a test with a sensitivity and specificity of over 90%, respectively. Recommended (see discussion) positive likelihood ratio parameter of >10 and negative likelihood ratio parameter of <0.1 are met with a score of 47 (A) and 43 (B), respectively.
Acknowledgments
Grant Support: This study was supported in part by NCRR grant M01-RR00425 to the Cedars-Sinai General Research Center Genotyping core; NIH/NIDDK grant P01-DK046763; the the Southern California Diabetes Endocrinology Research Center grant, DK063491; Cedars-Sinai Medical Center Inflammatory Bowel Disease Research Funds; The Feintech Family Chair in IBD (S.R.T.); The Abe and Claire Levine Chair in Pediatric IBD (M.D.) and The Cedars-Sinai Board of Governors’ Chair in Medical Genetics (J.I.R.). Additional funding through grants DK76984 (M.D.) and DK084554 (M.D. and D.P.B.M). The CHS research reported in this article was supported by contract numbers N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133; grant numbers U01 HL080295 and R01 HL087652 from the NHLBI, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/pi.htm.
Footnotes
Disclosures: Authors have no disclosures to declare. No conflicts of interest exist.
References
- 1.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–5. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 2.Fisher SA, Tremelling M, Anderson CA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet. 2008;40:710–2. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 4.Silverberg MS, Cho JH, Rioux JD, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–20. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson CA, Massey DC, Barrett JC, et al. Investigation of Crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology. 2009;136:523–9. doi: 10.1053/j.gastro.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asano K, Matsushita T, Umeno J, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325–9. doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 7.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farmer RG, Easley KA, Rankin GB. Clinical patterns, natural history, and progression of ulcerative colitis. A long-term follow-up of 1116 patients. Dig Dis Sci. 1993;38:1137–46. doi: 10.1007/BF01295733. [DOI] [PubMed] [Google Scholar]
- 9.Henriksen M, Jahnsen J, Lygren I, et al. Ulcerative colitis and clinical course: results of a 5-year population-based follow-up study (the IBSEN study) Inflamm Bowel Dis. 2006;12:543–50. doi: 10.1097/01.MIB.0000225339.91484.fc. [DOI] [PubMed] [Google Scholar]
- 10.Hendriksen C, Kreiner S, Binder V. Long term prognosis in ulcerative colitis--based on results from a regional patient group from the county of Copenhagen. Gut. 1985;26:158–63. doi: 10.1136/gut.26.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraga XF, Vergara M, Medina C, et al. Effects of smoking on the presentation and clinical course of inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1997;9:683–7. doi: 10.1097/00042737-199707000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Mokbel M, Carbonnel F, Beaugerie L, et al. Effect of smoking on the long-term course of ulcerative colitis. Gastroenterol Clin Biol. 1998;22:858–62. [PubMed] [Google Scholar]
- 13.Basu D, Lopez I, Kulkarni A, et al. Impact of race and ethnicity on inflammatory bowel disease. Am J Gastroenterol. 2005;100:2254–61. doi: 10.1111/j.1572-0241.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen GC, Torres EA, Regueiro M, et al. Inflammatory bowel disease characteristics among African Americans, Hispanics, and non-Hispanic Whites: characterization of a large North American cohort. Am J Gastroenterol. 2006;101:1012–23. doi: 10.1111/j.1572-0241.2006.00504.x. [DOI] [PubMed] [Google Scholar]
- 15.Roussomoustakaki M, Satsangi J, Welsh K, et al. Genetic markers may predict disease behavior in patients with ulcerative colitis. Gastroenterology. 1997;112:1845–53. doi: 10.1053/gast.1997.v112.pm9178675. [DOI] [PubMed] [Google Scholar]
- 16.Bouma G, Crusius JB, Garcia-Gonzalez MA, et al. Genetic markers in clinically well defined patients with ulcerative colitis (UC) Clin Exp Immunol. 1999;115:294–300. doi: 10.1046/j.1365-2249.1999.00797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Concha EG, Fernandez-Arquero M, Lopez-Nava G, et al. Susceptibility to severe ulcerative colitis is associated with polymorphism in the central MHC gene IKBL. Gastroenterology. 2000;119:1491–5. doi: 10.1053/gast.2000.20258. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto-Furusho JK, Uscanga LF, Vargas-Alarcon G, et al. Clinical and genetic heterogeneity in Mexican patients with ulcerative colitis. Hum Immunol. 2003;64:119–23. doi: 10.1016/s0198-8859(02)00772-3. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad T, Armuzzi A, Neville M, et al. The contribution of human leucocyte antigen complex genes to disease phenotype in ulcerative colitis. Tissue Antigens. 2003;62:527–35. doi: 10.1046/j.1399-0039.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez L, Nunez C, Mendoza JL, et al. A recombined haplotype in the major histocompatibility region contains a cluster of genes conferring high susceptibility to ulcerative colitis in the Spanish population. Inflamm Bowel Dis. 2005;11:785–91. doi: 10.1097/01.mib.0000179210.96025.23. [DOI] [PubMed] [Google Scholar]
- 21.Brant SR, Panhuysen CI, Nicolae D, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–92. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho GT, Gaya DR, Satsangi J. Multidrug resistance (MDR1) gene in inflammatory bowel disease: a key player? Inflamm Bowel Dis. 2005;11:1013–9. doi: 10.1097/01.mib.0000186488.53493.c3. [DOI] [PubMed] [Google Scholar]
- 23.Ho GT, Soranzo N, Nimmo ER, et al. ABCB1/MDR1 gene determines susceptibility and phenotype in ulcerative colitis: discrimination of critical variants using a gene-wide haplotype tagging approach. Hum Mol Genet. 2006;15:797–805. doi: 10.1093/hmg/ddi494. [DOI] [PubMed] [Google Scholar]
- 24.Weedon MN, McCarthy MI, Hitman G, et al. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med. 2006;3:e374. doi: 10.1371/journal.pmed.0030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, Elston RC. Using the optimal receiver operating characteristic curve to design a predictive genetic test, exemplified with type 2 diabetes. Am J Hum Genet. 2008;82:641–51. doi: 10.1016/j.ajhg.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans DM, Visscher PM, Wray NR. Harnessing the information contained within Genome-wide Association Studies to improve individual prediction of complex disease risk. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- 27.Henckaerts L, Van Steen K, Verstreken I, et al. Genetic risk profiling and prediction of disease course in Crohn’s disease patients. Clin Gastroenterol Hepatol. 2009;7:972–980. e2. doi: 10.1016/j.cgh.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Dubinsky M, Ling M, Friedman M, et al. Genome Wide Association (GWA) Predictors Of Anti-Tnfα Therapeutic Responsiveness In Pediatric Inflammatory Bowel Disease (IBD) Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lennard-Jones JE, Ritchie JK, Zohrab WJ. Proctocolitis and Crohn’s disease of the colon: a comparison of the clinical course. Gut. 1976;17:477–82. doi: 10.1136/gut.17.6.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 31.Psaty BM, O’Donnell CJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of Prospective Meta-Analyses of Genome-Wide Association Studies From 5 Cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gunderson KL, Steemers FJ, Ren H, et al. Whole-genome genotyping. Methods Enzymol. 2006;410:359–76. doi: 10.1016/S0076-6879(06)10017-8. [DOI] [PubMed] [Google Scholar]
- 33.Gunderson KL, Steemers FJ, Lee G, et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–54. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 36.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 37.R: A language and environment for statistical computing. Version 2.9.0. Vienna, Austria: 2006. [Google Scholar]
- 38.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–44. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 39.Lasko TA, Bhagwat JG, Zou KH, et al. The use of receiver operating characteristic curves in biomedical informatics. J Biomed Inform. 2005;38:404–15. doi: 10.1016/j.jbi.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Sing T, Sander O, Beerenwinkel N, et al. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–1. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 41.Efron B. Bootstrap Methods: Another look at the jackknife. The Annals of Statistics. 1979;7:1–26. [Google Scholar]
- 42.Zhu H, Rohwer R. No free lunch for cross-validation. Neural Computation. 1996;8:1421–1426. [Google Scholar]
- 43.Solberg IC, Lygren I, Jahnsen J, et al. Clinical course during the first 10 years of ulcerative colitis: results from a population-based inception cohort (IBSEN Study) Scand J Gastroenterol. 2009;44:431–40. doi: 10.1080/00365520802600961. [DOI] [PubMed] [Google Scholar]
- 44.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–40. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrett JC, Lee JC, Lees CW, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–4. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takedatsu H, Michelsen KS, Wei B, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–67. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 48.Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–47. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 49.McGovern DPB, Summerskill WSM, McManus R, et al. Evidence-Based Medicine in General Practice. BIOS Scientific Publishers Ltd; 2001. [Google Scholar]
- 50.Matanis T, Akhmanova A, Wulf P, et al. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat Cell Biol. 2002;4:986–92. doi: 10.1038/ncb891. [DOI] [PubMed] [Google Scholar]
- 51.Moorhead AR, Rzomp KA, Scidmore MA. The Rab6 effector Bicaudal D1 associates with Chlamydia trachomatis inclusions in a biovar-specific manner. Infect Immun. 2007;75:781–91. doi: 10.1128/IAI.01447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ide N, Hata Y, Nishioka H, et al. Localization of membrane-associated guanylate kinase (MAGI)-1/BAI-associated protein (BAP) 1 at tight junctions of epithelial cells. Oncogene. 1999;18:7810–5. doi: 10.1038/sj.onc.1203153. [DOI] [PubMed] [Google Scholar]
- 53.van Bodegraven AA, Curley CR, Hunt KA, et al. Genetic variation in myosin IXB is associated with ulcerative colitis. Gastroenterology. 2006;131:1768–74. doi: 10.1053/j.gastro.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 54.McGovern DP, Taylor KD, Landers C, et al. MAGI2 genetic variation and inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:75–83. doi: 10.1002/ibd.20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–7. doi: 10.1136/gut.2007.133132. [DOI] [PubMed] [Google Scholar]
- 56.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ogawa K, Tanaka K, Ishii A, et al. A novel serum protein that is selectively produced by cytotoxic lymphocytes. J Immunol. 2001;166:6404–12. doi: 10.4049/jimmunol.166.10.6404. [DOI] [PubMed] [Google Scholar]
- 59.Kuepper M, Bratke K, Julius P, et al. Increase in killer-specific secretory protein of 37 kDa in bronchoalveolar lavage fluid of allergen-challenged patients with atopic asthma. Clin Exp Allergy. 2005;35:643–9. doi: 10.1111/j.1365-2222.2005.02238.x. [DOI] [PubMed] [Google Scholar]
- 60.Yang H, Rotter JI, Toyoda H, et al. Ulcerative colitis: a genetically heterogeneous disorder defined by genetic (HLA class II) and subclinical (antineutrophil cytoplasmic antibodies) markers. J Clin Invest. 1993;92:1080–4. doi: 10.1172/JCI116613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stokkers PC, Reitsma PH, Tytgat GN, et al. HLA-DR and -DQ phenotypes in inflammatory bowel disease: a meta-analysis. Gut. 1999;45:395–401. doi: 10.1136/gut.45.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 63.Picornell Y, Mei L, Taylor K, et al. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–8. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tremelling M, Berzuini C, Massey D, et al. Contribution of TNFSF15 gene variants to Crohn’s disease susceptibility confirmed in UK population. Inflamm Bowel Dis. 2008;14:733–7. doi: 10.1002/ibd.20399. [DOI] [PubMed] [Google Scholar]
- 65.Thiebaut R, Kotti S, Jung C, et al. TNFSF15 polymorphisms are associated with susceptibility to inflammatory bowel disease in a new European cohort. Am J Gastroenterol. 2009;104:384–91. doi: 10.1038/ajg.2008.36. [DOI] [PubMed] [Google Scholar]
- 66.Bamias G, Martin C, 3rd, Marini M, et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–74. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 67.Prehn JL, Mehdizadeh S, Landers CJ, et al. Potential role for TL1A, the new TNF-family member and potent costimulator of IFN-gamma, in mucosal inflammation. Clin Immunol. 2004;112:66–77. doi: 10.1016/j.clim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 68.Zini N, Lisignoli G, Solimando L, et al. IL1-beta and TNF-alpha induce changes in the nuclear polyphosphoinositide signalling system in osteoblasts similar to that occurring in patients with rheumatoid arthritis: an immunochemical and immunocytochemical study. Histochem Cell Biol. 2003;120:243–50. doi: 10.1007/s00418-003-0563-y. [DOI] [PubMed] [Google Scholar]
- 69.McGraw DW, Almoosa KF, Paul RJ, et al. Antithetic regulation by beta-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway beta-agonist paradox. J Clin Invest. 2003;112:619–26. doi: 10.1172/JCI18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi GP, Bryant RA, Riese R, et al. Role for cathepsin F in invariant chain processing and major histocompatibility complex class II peptide loading by macrophages. J Exp Med. 2000;191:1177–86. doi: 10.1084/jem.191.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The clinical utility of the MR-UC risk score is demonstrated by positive (A) and negative (B) likelihood ratios; sensitivity (C) and specificity (D); and positive (E) and negative (F) predictive values. A score of 44 (C) and 47 (D) can be used to generate a test with a sensitivity and specificity of over 90%, respectively. Recommended (see discussion) positive likelihood ratio parameter of >10 and negative likelihood ratio parameter of <0.1 are met with a score of 47 (A) and 43 (B), respectively.