

Abstract
The classical allergic reaction starts seconds or minutes after Ag contact and is committed by Abs produced by a special subset of B lymphocytes. These Abs belong to the IgE subclass and are responsible for Type I hyperreactivity reactions. Treatment of allergic diseases with humanized anti-IgE Abs leads primarily to a decrease of serum IgE levels. As a consequence, the number of high-affinity IgE receptors on mast cells and basophils decreases, leading to a lower excitability of the effector cells. The biological mechanism behind anti-IgE therapy remains partly speculative; however, it is likely that these Abs also interact with membrane IgE (mIgE) on B cells and possibly interfere with IgE production. In the present work, we raised a mouse mAb directed exclusively against the extracellular membrane-proximal domain of mIgE. The interaction between the monoclonal anti-mIgE Ab and mIgE induces receptor-mediated apoptosis in vitro. Passive immunization experiments lead to a block of newly synthesized specific IgEs during a parallel application of recombinant Bet v1a, the major birch pollen allergen. The decrease of allergen-specific serum IgE might be related to tolerance-inducing mechanisms stopping mIgE-displaying B cells in their proliferation and differentiation.
The IgE Abs against normally innocuous environmental allergens are the key effector molecules in allergic diseases. In sensitized atopic individuals, allergen exposure induces cross-linking of high-affinity FcεRI receptor-bound IgE on effector cells and, thus, immediate release of anaphylactogenic mediators (1). Like other immunoglobulins, IgE consists of two light and two heavy chains (ε-isotype) and can be produced in two forms by alternative splicing: a secreted and a membrane-bound form. Membrane IgE (mIgE)4 is a transmembrane protein that behaves like a classical Ag receptor on B lymphocytes (2). Previous reports showed that the expression of a functional membrane form is essential for generating humoral isotype-specific IgE and IgG1 responses in mice and an IgG2 response in man (3-5). We concluded that the Ag receptor is the only device for an effective Ag presentation and that signals generated by the receptor are needed not only for the maturation process but also for the expansion of Ag-specific B cells. Ag binding by the Ag receptor is considered the first signal in B cell activation. Without a second signal, consisting of physical contact with the Ag-specific Th cell and the interleukins it produces, the activation is abortive. The abortively activated B cells disappear, either by an apoptotic mechanism or after a short anergic state, in which the B cells cannot be further activated or produce Ab (6-9). We therefore hypothesized that targeting mIgE without supplying a second signal should be a reasonable approach for interfering with IgE production in vivo (10) and considered the 19 amino acid isotype-specific extracellular membrane-proximal domain (EMPD) as an ideal target for the interference.
In the present work, we present a systemic approach, based on the isotype-specific targeting of mIgE-expressing cells with a mAb raised against the EMPD region of mIgE (Fig. 1). We show that targeting of mIgE by passive immunization indeed leads to a block of newly synthesized specific IgEs in vivo and induces apoptosis in vitro.
FIGURE 1.
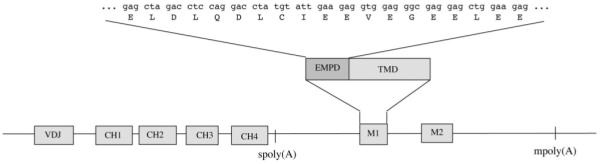
Like all other immunoglobulins, IgE can be expressed as secreted Ab or as membrane-anchored receptor. The membrane form of IgE is coded by the variable domain exon, 4 constant domain exons (CH1-CH4), the transmembrane domain exon M1, and the cytoplasmic domain exon M2. The 19 amino acids of the EMPD, coded by M1, were used as target sequence for the generation of anti-EMPD specific Abs.
Materials and Methods
Immunization and generation of hybridomas
To circumvent tolerance phenomenon, female ΔM1M2 BALB/c mice (11), unable to express membrane-bound IgE due to the lack of the IgE-transmembrane and IgE-cytoplasmic domains, were immunized with keyhole limpet hemocyanin (KLH)-coupled EMPD-peptide of mouse (KLH-ELDLQDLCIEEVEGEELEE) (synthesized by INBIOS). The KLH-EMPD peptide was adsorbed to Alum and injected intranodaly (12). In parallel, a 6 times repeated, recombinant EMPD protein (6xEMPD; HHHHHH (ELDLQDLCIEEVEGEELEE)6), cloned in vector pHIS2 and expressed as N-terminally [His]6-tagged protein in Escherichia coli was used for i.p. booster immunization (13).
A total of 0.5 μg of the EMPD-KLH conjugate was injected every 2 wk (until day 56) into the left inguinal lymph node of each mouse. After five times, to enhance the immune response (13), 10 μg of 6xEMPD was injected i.p. on day 66 and day 72. Subsequently, the mice were boosted with 10 μg EMPD-KLH i.p. three times (days 79, 81, and 86).
To generate hybridomas, lymph node and spleen cells of anti-EMPD positive mice were harvested and fused with the mouse B-lymphoma cell line Ag-8 (standard procedure). Culture supernatants were tested for anti-EMPD Abs by ELISA. Positive clones were expanded by limiting dilution to obtain specific monoclonal hybridomas.
Purification of the mAb
Anti-EMPD-specific hybridomas were grown in tissue culture flasks (Greiner Bioscience) for 14 days and finally centrifuged at 13,000 g at 4°C for 30 min. A total of 150 ml supernatant was concentrated with centriprep50 columns to 20 ml total volume and applied to an anti-mouse IgG1-agarose column (Sigma-Aldrich), previously equilibrated with 20 mM NaPO4 binding buffer. Elution was performed with 0.1 M glycine HCl (pH 2.7). For pH-neutralization 100 μl 1M Tris-HCl (pH 8.1) was added to each fraction. The fractions were tested for their IgG1 content under reducing and nonreducing conditions in Coomassie-staining and Western blot with AP-labeled goat anti-mouse IgG1. Positive fractions were pooled and dialyzed against PBS with 0,05% NaN3. The protein concentration was measured with BCA-assay kit (Pierce).
Sequence analysis
RNA of the hybridoma cells was isolated with an RNeasy kit (Promega), followed by cDNA synthesis with first strand reaction beads and oligo(dT)-primer. DNA amplification was performed with a mix of VH primers (VHa: [5′-gaggttcagctgcagcag(ct)c-3′]; and VHb: [5′-gaggtgcagctggtgga(ag)tc-3′]), a constant reverse primer for γ1 H chain (CH2γ1: [5′-ttaggagtcagagtaatggtgagcacatcc-3′]), a mix of VL primers (Vκ1: [5′-gatgttttgatgacccaaactcca-3′]; Vκ4: [5′-caaattgttctcacccagtctcca-3′]; Vκ10: [5′-gatatccagatgacacagactaca-3′]; and Vκ24: [5′-gatattgtgatgacgcaggctgca-3′]), and a constant κ L chain reverse primer ([5′-gatggatacagttggtgc-3′]).
The γ1 chains were cloned into pCR-script Amp vector (Qiagen). Analysis of the sequence was performed with the ABI-PRISM.. sequencing kit of Applied Biosystems using M13 forward [5′-gtaaaacgacggccagt-3′] and reverse [5′-ggaaacagctatgaccatg-3′] primers.
Determination of the binding affinity by surface plasmon resonance (Biacore X)
Recombinant His-tagged 6xEMPD (250 μg/ml in PBS) was diluted 1/1 in 10 mM NaAcetate (pH 4) and coupled to a CM5 chip according to the manufacturer’s instructions. Approximately 1700 resonance units rec. 6xEMPD-His were coupled to flow cell 1. Empty flow cell 2 served as a reference. mAbA9 was injected at different concentrations (60 nM-6.2 μM) in HBS-EP buffer (Biacore) and the surface plasmon resonance was recorded. Data were analyzed with BIAevaluation software (Biacore).
ELISA
Competition ELISA
Nunc-96-well plates were coated with 50 ng 6xEMPD per well. mAbA9 was preincubated with increasing amounts of 6xEMPD peptide from 10 to 100 molar excess. After 2 h, the preincubated Ab-peptide complex was added to the 6xEMPD coated plate followed by incubation for 2 h at room temperature (RT). As secondary Ab AP-labeled goat anti-mouse IgG1 (1: 3000 in PBS) was incubated at RT for 1 h. After development with AP-substrate, the absorption was measured at 405–492 nm. Data were expressed as % competition against 6xEMPD concentration.
Total IgE ELSA
Coating was done with 500 ng/100 μl per well of anti-IgE 84.1C in PBS overnight After blocking with PBS containing 1% BSA (1 h, 37°C) plates were washed with PBS and mice sera were added in dilutions 1/20, 1/60, and 1/180 in PBS/0.1% BSA at RT for 2 h. As standard, purified mouse IgE was used. As detection Ab, AP-labeled rat anti-mouse IgE (Southern Biotechnology Associates), diluted at 1 μg/ml in PBS/0.1% BSA, was used. The absorption was measured at 405–492 nm.
Specific IgE ELISA
For Ag-specific ELISA measurements, Nunc-96-well plates were coated with 500 ng recombinant Bet v1a/well. Sera were diluted 1/10, 1/30, and 1/90 in PBS containing 0.1% BSA added to the plate and incubated at RT for 2 h. For detection, AP-labeled rat anti-mouse IgE (Southern Biotechnology Associates) was added at a concentration of 1 μg/ml in PBS/0.1% BSA and plates were incubated at RT for 2 h. Absorption measurement was read out at 405–492 nm. Titers were expressed as the reciprocal serial dilution, at which a half-maximal OD value was measured.
FACS analysis
K46 mIgE+ cells were incubated with 500 ng of purified FITC-mAbA9 and/or FITC-IgE (R35–72). Appropriate isotype controls were used.
Rat basophilic lymphocytes (RBL)-assay
RBLs were plated to 4 × 104 cells/well in a volume of 100 μl RPMI 1640-RBL medium in flat-bottom wells tissue culture plate and incubated overnight at 37°C and 7.5% CO2. On the next day, 50 μl supernatant was removed and serum dilutions ranging from 1/20 to 1/320 were added in prewarmed RPMI 1640. Plates were further incubated for 2 h at 37°C followed by two washes with 200 μl tyrode/BSA wash buffer. For stimulation, 5 μg Bet v1a, anti-IgE Ab EM95-3, 84-1C, or mAbA9 were added at different concentrations. Cells were then incubated again for 30 min at 37°C. For a 100% release of β-hexosaminidase, cells were lysed by adding 1% Triton X-100. Plates were then centrifuged for 5 min, 260 g at RT. Afterward, 50 μl supernatant was transferred to a new 96-well. A total of 50 μl assay solution (0.1M citric acid (pH 4.5) and 80 μl 10 mM 4-methylumbelliferyl N-acetyl-β-D-glucosaminide) was added and the plates were incubated at 37°C and 7.5% CO2 for 1 h. The reaction was stopped by addition of 100 μl glycine (pH 10.7). The fluorescence (SpectraFlour) was measured at the bottom of the plate (excitation 360 nm; emission 465 nm). Measured values were converted to % of total release obtained by triton lysis.
CaspACE FITC-VAD-FMK apoptosis assay
mIgE-positive K46 cells were incubated with mAbA9 at a concentration of 0.3 μg/ml in a total volume of 1 ml in RPMI 1640/7.5% FCS in flat-bottom 48-well plates (Greiner Bioscience). After 6, 24, and 30 h, the percentage of apoptotic cells was measured using the CaspACE FITC-VAD-FMK (Promega) apoptosis assay, according to manufacturer’s instructions.
Results
Generation of anti-EMPD-specific hybridomas
mIgE-EMPD-specific hybridomas were raised in the ΔM1M2 mouse strain (11) to avoid tolerance observed in mice expressing a functional mIgE-bearing BCR. The administration through the intralymphatic route was followed by i.p. booster immunization to enhance the immune response (12, 13). The increase in specific anti-EMPD titer was determined by ELISA. At days 77 and 85, two of the five mice showed high specific IgG1 titers. The spleens and lymph nodes of these two mice were used for generation of hybridomas. Ten single hybridomas were identified as anti-EMPD secreting clones of IgG1 isotype. Clone A9 was selected for production and purification of the corresponding mAb (mAbA9).
Cloning and sequencing of H and L chains of mAbA9
We next cloned and sequenced mAbA9. After isolation of total mRNA and first strand cDNA synthesis, the H and L chains were amplified with primer mixes for VH and VL variable region and specific constant region primers. Amplicons were cloned into the pCR-Script and sequenced. Blast search identified the variable heavy region of anti-EMPD γ1-chain as a member of the V11 germline gene family and the variable region of the κ L chain as a member of the kj4 germline gene family. Variable H and L chain sequences of the mAbA9 Ab were submitted to GenBank (accession numbers: EF156450 for the VH and EF156451 for VL).
mAbA9 binds the EMPD-region with high specificity and affinity
To test the specificity of mAbA9 for 6xEMPD, competition-ELISAs were performed. Competition was achieved by preincubation of mAbA9 with increasing amounts of purified recombinant 6xEMPD peptide. With 10-fold molar excess of 6xEMPD peptide 43% competition was measured, whereas 87% inhibition was obtained with 100 molar excess. No competition was observed with soluble mouse IgE, indicating specificity of mAbA9 for the 6xEMPD peptide (Fig. 2a).
FIGURE 2.

The specificity and affinity of mAbA9 for the EMPD region was determined with inhibition ELISA and SPRA. a, Competitive inhibition ELISA: mAbA9 was preincubated with increasing amounts of 6xEMPD peptide (from 10 to 100 molar 6xEMPD excess). No competition was achieved with soluble mouse IgE. Values are means ± SD of five inhibition experiments. b, Determination of the binding affinity: SPRA-analysis was performed, by injecting increasing amounts of mAbA9. Relative response units served as data points for the subsequent scatchard plot analysis. The slope of the deduced straight line equation was used to determine the binding affinity constant.
The association between the recombinantly expressed His-tagged 6xEMPD peptide and mAbA9 was tested by surface plasmon resonance analysis (SPRA) using a Biacore X device. A total of 1700 resonance units of recombinant His-tagged 6xEMPD were coupled to flow cell 1 of a CM5 chip, empty flow cell 2 served as reference. The purified mAbA9 was injected at different concentrations (60 nM-6.2 μM), and the surface plasmon resonance was recorded. The Kd of 7.4 × 10−9 M, determined by scatchard plot analysis from the respective curves (Fig. 2b), shows a high affinity of mAbA9 for the substrate.
mAbA9 recognizes surface mIgE
To demonstrate the capacity of mAbA9 to bind surface expressed mIgE, cell line K46 mIgE+ was incubated with purified FITC-labeled mAbA9 and with FITC-labeled rat anti-soluble mouse IgE (clone R35–72), respectively. To define lower cut off values, FITC-labeled isotype controls for mouse IgG1 and rat IgG1 were used. Additionally, we stained the untransfected cell line K46 with FITC-labeled mAbA9 as control. The FACS analysis demonstrated that mAbA9 specifically recognizes mIgE and underlines the capacity of mAbA9 to recognize labeled surface expressed mIgE (Fig. 3).
FIGURE 3.
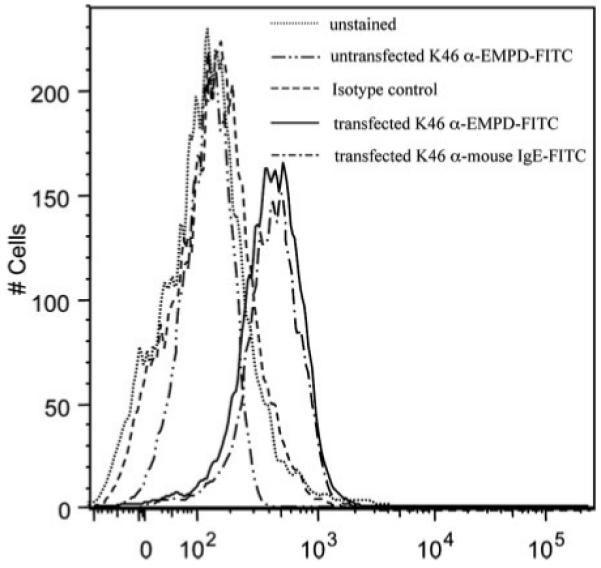
To demonstrate the capacity of mAbA9 to bind surface expressed mIgE, FACS analysis was performed. FITC-labeled mAbA9 and rat anti-mouse soluble IgE recognize mIgE on cell line K46 mIgE+ in a comparable way. The isotype control as well as the untransfected cell line stained with FITC-labeled mAbA9 overlap with unstained cells.
mAbA9 does not interact with soluble IgE bound to FcεRI
To demonstrate that mAbA9 does not interact with soluble IgE bound to FcεRI, RBL cells were cultivated and incubated with serially diluted serum samples of Bet v1a-immunized mice. Total cell lysis of the RBL cells with Triton X-100 led to a complete release of the basophil-specific enzyme β-hexosaminidase and served as control (100%). Addition of Bet v1a Ag led to a release of 7.6%, indicating the specificity of the IgE Abs for Bet v1a. Rat anti-mouse IgE Ab EM95-3 and 84-1C induced a release of 27.8 and 17%, respectively. As expected, incubation of receptor-bound IgE with two concentrations of mAbA9 did not induce any relevant release of β-hexosaminidase (1.8 and 2%). These results show that mAbA9 is a nonanaphylactic anti-IgE Ab unable to cross-link receptor-bound IgE molecules (Fig. 4).
FIGURE 4.
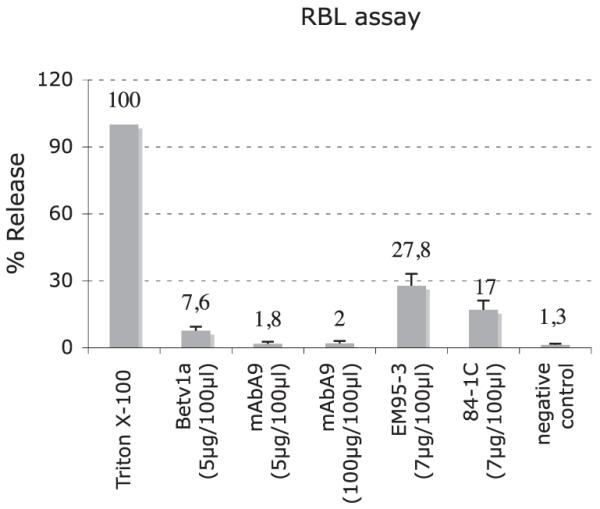
To demonstrate that mAbA9 fulfils the criteria of a non-anaphylactogenic Ab, RBL cells were incubated with serum of Bet v1a-immunized mice and subsequently stimulated with Bet v1a, mAbA9, and two different anti-IgE Abs (EM95-3 and 84-1C). Values are means ± SD of three degranulation assays.
Passive administration of mAbA9 affects the specific serum IgE levels
We next asked whether a passive administration of mAbA9 affects the specific serum IgE levels. Therefore, mice of control group 1 were immunized with the allergen Bet v1a (20 μg/immunization i.p.) on days 0, 7, 14, and 183. Group 2 was immunized as described for group 1, but additionally received monoclonal anti-EMPD Ab on days 3, 6, 9, 12, and 15. The “3-day scheme” reflects the half-life of IgG1 in mice. In previous publications (11, 14), we outlined the necessity of sera depletion for the IgG fraction when performing a specific IgE ELISA. However, Bet v1a represents a relatively weak Ag in the mouse if compared with OVA. The increase of specific IgG1s during the whole course of immunization only spans over 2 decades (Fig. 5b). In a test ELISA measurement, no difference between IgG depleted and undepleted sera was observed (data not shown). We therefore decided to perform all Bet v1a specific IgE ELISA measurements with undepleted sera. The specific Bet v1a titer in the control group continually increased during the course of immunization and reached a first peak at day 21. As described earlier (14), specific IgE titers decreased rapidly. However, a clear memory response to Bet v1a was detectable after a booster immunization (day 183) at day 190. In contrast, parallel application of mAbA9 strongly repressed Bet v1a-specific IgE titers. At day 21, the allergen-specific IgE titer was decreased by 64%. Notably, group 2 did not receive mAbA9 anymore after day 15, yet a strong suppression of allergen-specific IgE was still present at day 190 as reflected by the 74% decrease of the Bet v1a titer compared with the control group (Fig. 5a). These data are compatible with a missing memory response due to the depletion of IgE-specific Bet v1a memory B cells: newly developing IgE-secreting plasma cells have to go through an mIgE-expressing B cell stage during differentiation and their generation is abrogated by anti-mIgE treatment. This assumption is corroborated by the fact that the Bet v1a-specific IgG1 Ab response was not affected by passive immunization with mAbA9. No differences in the IgG1 titers between control and experimental groups could be observed during the course of immunization (Fig. 5b). Mean values of the different groups are expressed as closed (group 1) or dotted (group 2) lines. Differences between the groups for time point d21 and d190 for specific IgE were significant (p ≤ 0.01). For calculation of the p values, the paired t test was used.
FIGURE 5.

To show that mAbA9 blocks specific IgE synthesis during a passive immunization approach, mice of control group 1 (open circles) were immunized with the allergen Bet v1a on days 0, 7, 14, and 183. Group 2 (closed circles) was immunized as described for group 1 but additionally received monoclonal anti-EMPD Ab on days 3, 6, 9, 12, and 15. Each circle represents one individual mouse. Bet v1a-specific IgE (a) and IgG1 (b) titers were determined.
mAbA9 passive administration does not affect the total IgE level
We next asked whether the total IgE level was also affected in mAbA9-immunized mice. Groups 1 and 2 were tested for their total IgE serum levels. During the course of immunization, total IgE levels weakly increased from 1 μg/ml up to 2.5 μg/ml, without any significant difference between the investigated groups (Fig. 6a). We conclude that mAbA9 has no effect on total IgE levels, and we assume that IgE without specificity for Bet v1a is produced by pre-existing long living plasma cells (15-17), which are not a target for mAbA9 because they do not express mIgE on their surface (16), but reside in survival niches in the bone marrow (15, 18-20). Alternatively, total IgE levels may result from cells that are recruited in the IgE response after treatment or by cells that produce IgE in an IL-4-dependent, but further T cell independent way (21).
FIGURE 6.

Although specific IgE titers were successfully repressed, mAbA9 had no effect on already established total IgE titers. a, Total IgE levels of group 1 (open circles) and group 2 (closed circles) during the course of immunization. Mean values of the different groups are expressed as closed (group 1) or interrupted (group 2) lines. Differences between the groups for time point d21 and d190 for specific IgE were significant (p ≤ 0.01). For calculation of the p values the paired t test was used. b, Specific IgE titers of mAbA9-pretreated mice (closed circles) in comparison with untreated mice.
Compatible with the data described above, we finally showed experimentally that already established specific IgE responses couldn’t be reduced by subsequent administration of mAbA9. Group 3 (open circles) was immunized with Bet v1a on days 0, 7, and 14 and thus served as control. Group 4 (closed circles) additionally received mAbA9 at days 18, 21, 24, 27, and 30. No difference in the Bet v1a specific IgE titer between the two groups was observed (Fig. 6b), indicating that already established specific IgE titers cannot be suppressed with mAbA9 in a passive immunization approach.
mAbA9 induces apoptosis in B cells expressing mIgE in vitro
We finally tested whether receptor-mediated stimulation can induce apoptosis of mIgE-expressing B cells in vitro. mIgE-transfected K46 mouse lymphoma cells were stimulated with mAbA9. Subsequent FACS analysis showed a time-dependent increase of an apoptotic cell population compared with unstimulated cells (Fig. 7). After 6 h, an apoptotic cell population of 19% could be detected. After 24 h, the maximum of 36% apoptotic cells was reached followed by a decrease of apoptotic cells to 27% after 30 h. As control, unstimulated cells after 24 h were used. This result demonstrates that mAbA9 induces apoptosis in a receptor mediated process.
FIGURE 7.
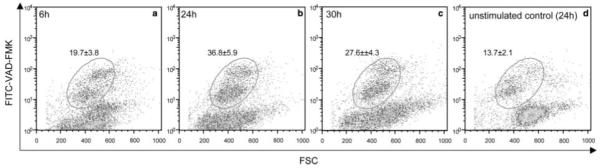
To investigate whether receptor-mediated stimulation can induce apoptosis of mIgE-expressing B cells in vitro, mIgE expressing cell line K46 was stimulated with mAbA9. Appearance of apoptotic cell populations was assessed in a time dependent (a–c) fashion using a CaspACE FITC-VAD-FMK apoptosis assay. Unstimulated cells after 24 h served as control (d). Values are means ± SD of three apoptosis assays.
Discussion
In the current dogma, the B cell binds the Ag via its receptor, which transduces the first signal in the cell via activation of tyrosine kinases, such as syk and lyn (reviewed in Ref. 22). In the following, the receptor-Ag complex gets internalized, processed, and displayed as peptide-MHC class II complex on the B cell surface. Ag binding to the B cell also stimulates expression of the costimulatory molecules B7-1 and B7-2. If T cells with receptors that can recognize these coreceptor-peptide-MHC complexes exist, then the ensuing T cell-B cell interaction provides the second signals in the form of a CD40 ligand-CD40 interaction and the release of stimulatory cytokines, such as IL-4 and IL-5 or IFN-γ. In this framework of thinking, Ag stimulation of B cells in the absence of T cell help leads to an abortive immune response (23), while stimulation in the presence of Ag-specific T cell help leads to proliferation and differentiation. We assume that this way of reasoning can be adopted to explain the induction of tolerance of a class-switched mIgE-bearing B cell population by using anti-IgE Abs.
The aim of our work was to develop mAbs able to specifically target membrane-bound IgE, which in the context of the BCR is indispensable for the production and quality of secreted IgE, and to test their ability to block IgE synthesis in vivo. mIgE is a splicing variant of soluble IgE that contains, beside a transmembrane region and a cytoplasmic tail, an isotype-specific 19-amino acid long EMPD (Fig. 1). We used a recombinantly produced EMPD multimeric fusion protein to immunize ΔM1M2 mice (11). This special mouse strain was used to avoid tolerance observed in mice expressing a functional mIgE bearing BCR. EMPD protein was administered through the intralymphatic route followed by i.p. booster immunization to enhance the immune response (13). Using this procedure, we were able to isolate a high affinity mouse monoclonal anti-mouse EMPD Ab of γ1-isotype termed mAbA9 (Fig. 2b). Specificity for the 19 amino acids of the IgE-EMPD region was demonstrated by ELISA, inhibition ELISA, and by the lack of binding to soluble IgE (Fig. 2a). FACS analysis of the mIgE-expressing K46 cell line was used to demonstrate the ability of the mAbA9 to recognize the native conformation of mIgE (Fig. 3). Additionally, we could demonstrate that even high concentrations of mAbA9 failed to induce degranulation of RBL cells (Fig. 4). At this stage, mAbA9 fulfilled all criteria required to be a promising lead compound for passive immunization experiments.
In a prophylactic study, mAbA9 was passively administrated to mice in parallel with Bet v1a as sensitizing allergen. Compared with the control group only treated with Bet v1a, we observed a drastic reduction in the production of Bet v1a-specific IgE Abs at day 21 (−64%) with a long lasting effect on the development of a Bet v1a-specific IgE-memory cell population. In fact, challenge of both mice groups with Bet v1a at day 183 quickly restored the Bet v1a-specific IgE titer levels at day 190, whereas the mAbA9 treated group still showed a marked reduction of the titers (74%) despite the fact that the administration of the anti-mIgE Ab was stopped at day 15 (Fig. 5a). Also, the subsequent immunization of Bet v1a/mAbA9 pretreated mice with a second Ag (Phl p5) was not impaired and exactly resembled a classical primary immune response (data not shown). The same is true for the suppression of an already established specific IgE response. Treatment of mice with mAbA9 at a particular time where specific Bet v1a IgEs already exist failed to induce a reduction of the specific IgE serum titer (Fig. 6b).
We conclude that a parallel administered passive immunization with mAbA9 strongly suppresses the development of an allergen-specific IgE response, whereas Ab responses of other isotypes are not influenced as exemplified for the Bet v1a-specific IgG1 response (Fig. 5b). Although mAbA9 induced strong apoptosis in vitro if added to mIgE-expressing K46 cells (Fig. 7), we cannot yet verify whether this mechanism is responsible for the suppression of memory B cell development in vivo. The extremely low number of mIgE-expressing B cells present in vivo (14, 24) hampers a direct, satisfactory FACS analysis of this population, which would represent a suitable method to show apoptosis events. However, supportive data concerning the apoptosis hypothesis were published by Poggainella et al. (25). Poggianella et al. (25) studied the role of the EMPD region of human mIgE upon BCR engagement with anti-Id Abs. They constructed chimeric EMPD deleted ε-chains, transfected these chains into the mouse cell line A20, and showed that this region is involved in controlling Ca2+ mobilization and apoptosis induction. Interestingly, human mIgE is expressed in two EMPD-isoforms (long and short EMPD). Only the short version can be found as homologous domain in the mouse, while the long version is missing. Poggianella et al. (25) showed in an in vitro approach that “long EMPD mIgEs” are apoptosis resistant, whereas EMPD deleted constructs as well as “short EMPD mIgEs” were sensitive for apoptosis induction. In our manuscript, we went one step further and showed in vivo that exclusively targeting the mIgE population with an anti-EMPD Ab dramatically reduces the amount of specific IgEs during a passive immunization approach. We focused on the mIgE specific targeting by the construction of a mAb, which exclusively recognizes the mouse mIgE-EMPD region. One explanation of the observed reduction might be the active elimination of newly formed mIgE B cells as shown by Poggianella et al. (25) after anti-Id Ab crosslinkage. We deduce from that result that in the murine system, mIgE expressing B cells, which exclusively express short EMPDs, are sensitive to undergo apoptosis. In the present manuscript, we make a claim on the possibility to specifically target the memory portion of IgE and show a proof of principal that anti-EMPD treatment might be a therapeutic alternative in the future.
Interestingly, the total serum IgE levels are not influenced by the mAbA9 treatment (Fig. 6a). Even a prolonged administration of mAbA9 from day 3 up to day 30 (data not shown) did not lead to a decrease of total IgE levels in direct answer to mAbA9. However, we were not surprised but rather expected that result. Plasma cells (15, 17) are the main providers for the circulating pool of Abs and thus for humoral immunological memory, which makes the plasma cell of foremost importance in immune defense. Most of them return to their “place of birth” and home to the bone marrow or inflamed tissues where they persist for up to several months to years in survival niches as resident, immobile cells (15, 18-20). Hofer et al. (18) showed that the natural turnover of plasma cells in the bone marrow would never lead to a complete loss of an already established specific “plasma memory” response. Because mAbA9 exclusively recognizes mIgE, plasma cells will not be targeted by mAbA9, and considering the longevity of plasma cells, it is not surprising that an already established IgE level based on “plasma memory” cells will not considerably decline during a mouse life span. It is true that our data only show an effect if mAbA9 is administrated during immunization. However, the therapeutic potential of the anti-mIgE therapy consist in the suppression of newly developing IgE responses to allergens present in extracts used for allergen-specific immunotherapy frequently observed in clinical practice and not in the suppression of already established IgE-specific immune responses. Therefore our concept was: keep serum IgE untouched but suppress newly developing IgE responses. Both prerequisites could be achieved with mAbA9.
From the presented data, we conclude that mAbA9 cross-links the Ag receptor and induces apoptosis, as demonstrated by in vitro experiments. Even in case of initial survival of the mIgE+ B cells, it has to be assumed that these B cells have been rendered anergic, shortening their lifespan considerably without participation in the immune response. In future investigations also, the way and time of administration of the anti-mIgE Abs have to be carefully chosen, as demonstrated by the conclusions of Le Gros et al. (26): they propose that in mice, in which a strong IgE response had been induced with anti-IgD Abs, a subsequent s.c. treatment with goat-anti-mouse IgE Abs resulted in activation of IgE-secreting memory B cells. However, a careful analysis of their data allows a different conclusion: s.c. anti-IgE treatment abolishes an Ag-specific IgE-response, but enhances the secretion of IgE Abs with unknown specificity. These could well be those IgE Abs that are produced in an IL-4-dependent, but further T cell independent, way.
Summarizing, targeting mIgE bearing B cells with anti-mIgE-specific Abs might be a therapeutic alternative in the future. The advantage of this therapeutic approach would be the next step in the generation of anti-IgE therapy, with the advantage of inhibiting IgE secretion before specific secreted IgE production starts.
Footnotes
This work was supported by the Austria Science Foundation Projects P-19017 and T166 (Hertha Firnberg Fellowship) and the Austrian National Bank (Oesterreichische Nationalbank Grant 11710), Christian Doppler Laboratory for Allergy Diagnosis and Therapy, the Swiss National Science Foundation (Grant 310000-114634/1), and the OPO-Pharma Foundation, Zürich.
- mIgE
- membrane IgE
- EMPD
- extracellular membrane-proximal domain
- KLH
- keyhole limpet hemocyanin
- RT
- room temperature
- RBL
- rat basophilic lymphocyte
- SPRA
- surface plasmon resonance analysis
Disclosures
The authors have no financial conflict of interest.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Sutton BJ, Gould HJ. The human IgE network. Nature. 1993;366:421–428. doi: 10.1038/366421a0. [DOI] [PubMed] [Google Scholar]
- 2.Venkitaraman AR, Williams GT, Dariavach P, Neuberger MS. The B-cell antigen receptor of the five immunoglobulin classes. Nature. 1991;352:777–781. doi: 10.1038/352777a0. [DOI] [PubMed] [Google Scholar]
- 3.Achatz G, Lamers MC. In vivo analysis of the cytoplasmic domain of mIgE antibodies. Int. Arch. Allergy Immunol. 1997;113:142–145. doi: 10.1159/000237529. [DOI] [PubMed] [Google Scholar]
- 4.Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, Akira S. IκB kinase α is essential for mature B cell development and function. J. Exp. Med. 2001;193:417–426. doi: 10.1084/jem.193.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terada T, Kaneko H, Fukao T, Tashita H, Li AL, Takemura M, Kondo N. Fate of the mutated IgG2 heavy chain: lack of expression of mutated membrane-bound IgG2 on the B cell surface in selective IgG2 deficiency. Int. Immunol. 2001;13:249–256. doi: 10.1093/intimm/13.2.249. [DOI] [PubMed] [Google Scholar]
- 6.Goodnow CC. B-cell tolerance. Curr. Opin. Immunol. 1992;4:703–710. doi: 10.1016/0952-7915(92)90049-k. [DOI] [PubMed] [Google Scholar]
- 7.Goodnow CC, Cyster JG, Hartley SB, Bell SE, Cooke MP, Healy JI, Akkaraju S, Rathmell JC, Pogue SL, Shokat KP. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 1995;59:279–368. doi: 10.1016/s0065-2776(08)60633-1. [DOI] [PubMed] [Google Scholar]
- 8.Nemazee D, Buerki K. Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc. Natl. Acad. Sci. USA. 1989;86:8039–8043. doi: 10.1073/pnas.86.20.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 10.Infuhr D, Crameri R, Lamers R, Achatz G. Molecular and cellular targets of anti-IgE antibodies. Allergy. 2005;60:977–985. doi: 10.1111/j.1398-9995.2005.00832.x. [DOI] [PubMed] [Google Scholar]
- 11.Achatz G, Nitschke L, Lamers MC. Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science. 1997;276:409–411. doi: 10.1126/science.276.5311.409. [DOI] [PubMed] [Google Scholar]
- 12.Johansen P, Haffner AC, Koch F, Zepter K, Erdmann I, Maloy K, Simard JJ, Storni T, Senti G, Bot A, et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur. J. Immunol. 2005;35:568–574. doi: 10.1002/eji.200425599. [DOI] [PubMed] [Google Scholar]
- 13.Maloy KJ, Erdmann I, Basch V, Sierro S, Kramps TA, Zinkernagel RM, Oehen S, Kundig TM. Intralymphatic immunization enhances DNA vaccination. Proc. Natl. Acad. Sci. USA. 2001;98:3299–3303. doi: 10.1073/pnas.051630798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luger E, Lamers M, Achatz-Straussberger G, Geisberger R, Infuhr D, Breitenbach M, Crameri R, Achatz G. Somatic diversity of the immunoglobulin repertoire is controlled in an isotype-specific manner. Eur. J. Immunol. 2001;31:2319–2330. doi: 10.1002/1521-4141(200108)31:8<2319::aid-immu2319>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 15.Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004;199:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 1998;10:1703–1711. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- 17.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 18.Hofer T, Muehlinghaus G, Moser K, Yoshida T, Mei HE, Hebel K, Hauser A, Hoyer B, Luger EO, Dorner T, et al. Adaptation of humoral memory. Immunol. Rev. 2006;211:295–302. doi: 10.1111/j.0105-2896.2006.00380.x. [DOI] [PubMed] [Google Scholar]
- 19.Muehlinghaus G, Cigliano L, Huehn S, Peddinghaus A, Leyendeckers H, Hauser AE, Hiepe F, Radbruch A, Arce S, Manz RA. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105:3965–3971. doi: 10.1182/blood-2004-08-2992. [DOI] [PubMed] [Google Scholar]
- 20.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 21.Katona IM, Urban JF, Jr., Kang SS, Paul WE, Finkelman FD. IL-4 requirements for the generation of secondary in vivo IgE responses. J. Immunol. 1991;146:4215–4221. [PubMed] [Google Scholar]
- 22.Geisberger R, Crameri R, Achatz G. Models of signal transduction through the B-cell antigen receptor. Immunology. 2003;110:401–410. doi: 10.1111/j.1365-2567.2003.01770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carsetti R, Kohler G, Lamers MC. A role for immunoglobulin D: interference with tolerance induction. Eur. J. Immunol. 1993;23:168–178. doi: 10.1002/eji.1830230127. [DOI] [PubMed] [Google Scholar]
- 24.Achatz G, Luger E, Geisberger R, Achatz-Straussberger G, Breitenbach M, Lamers M. The IgE antigen receptor: a key regulator for the production of IgE antibodies. Int. Arch. Allergy Immunol. 2001;124:31–34. doi: 10.1159/000053661. [DOI] [PubMed] [Google Scholar]
- 25.Poggianella M, Bestagno M, Burrone OR. The extracellular membrane-proximal domain of human membrane IgE controls apoptotic signaling of the B cell receptor in the mature B cell line A20. J. Immunol. 2006;177:3597–3605. doi: 10.4049/jimmunol.177.6.3597. [DOI] [PubMed] [Google Scholar]
- 26.Le Gros G, Schultze N, Walti S, Einsle K, Finkelman F, Kosco-Vilbois MH, Heusser C. The development of IgE+ memory B cells following primary IgE immune responses. Eur. J. Immunol. 1996;26:3042–3047. doi: 10.1002/eji.1830261233. [DOI] [PubMed] [Google Scholar]