

Abstract
Microglial cells elaborate trophic factors and cytokines and remove toxins and debris from the extracellular space, acting analogously to peripheral macrophages. Over the past two decades increased attention has been directed at the role of microglia, not only in normal physiology, but also in mediating neurotoxicity. Activation of microglia is inherent to multiple neurodegenerative disorders and exposure to toxic compounds. In large measure, these revelations have come about as a result of technologies that enable researchers to obtain high-yield and -purity primary cultures of rodent microglia. The mechanical isolation protocol discussed in this protocol offers an economical method to isolate large amount of microglia in a short and not too labor intensive manner. Most importantly, it assures a high yield of cells with great reproducibility. Given the ever increasing importance of microglia to the field of neurotoxicology research, the ability to isolate high yield of primary microglia makes it possible to investigate the role and mechanisms associated with microglial modulation of neurotoxicity. We provide a detailed description on the methods that is routinely used in our laboratory for the isolation and culturing of microglia, with emphasis on the steps which are deemed most critical for obtaining pure and healthy cultures.
Keywords: microglia, isolation, culture, in vitro
INTRODUCTION
Neurons are the major cell type in the central nervous system (CNS), their unique electrical excitability distinguishing them from other cell types. Glial cells, including astrocytes, microglia and oligodendrocytes are non-electrically excitable. The term glia is derived from the Greek language and means “glue”. Yet glial cells have diverse functions, well beyond the original scope ascribed to them as being the mere glue that holds the neurons in place. They provide support and nutrition (Hamilton, Hillard et al. 2007), maintain CNS homeostasis (Vernadakis 1988), form myelin (Vernadakis 1988) and participate in signal transmission (Allen and Barres 2005; Schwabe, Bainton et al. 2005), just to name a few of their functions. The majority of glial cells are astrocytes, which are intimately involved in maintaining the extracellular milieu. Some selected functions of astrocytes include secretion of growth factors, release of cytokines and modulation of synaptic transmission by removal of neurotransmitters such as glutamate from the synaptic cleft. Oligodendrocytes are the myelin-forming cells in the CNS and are analogues to Schwann cells of the peripheral nervous system.
Microglia comprise approximately 12% of total cell population in the brain and are the second largest glial cell type. Microglia are derived from myeloid lineage (Ransohoff and Perry 2009). In postnatal rodents, immunocytochemical studies using antibodies to F4/80 (ERM1), CR3 and FcγRII/III establish that the microglia enter the brain from the circulation and are derived from circulating monocytes (Ransohoff and Perry 2009). In the brain, they possess phagocytic activity and survey the parenchyma. Microglia express low level of Major Histocompatibility Complex (MHC-II) and function as antigen presenting cells (APC) of the CNS (Ransohoff and Perry 2009). They are in close contact with other cells in the brain, mainly astrocytes, thus establishing cross-talk that mediates and synergizes responses both in physiological and pathological conditions. Under optimal conditions microglial surface receptors, such as TREM2 and Siglecs maintain the microglia in an inactivated state (Ransohoff and Perry 2009). In the presence of inflammation, infection, trauma and degenerative diseases, microglia are the first cell type to become activated. Their activation is inherent to Alzheimer’s disease (Shie, Nivison et al. 2009), Parkinson’s disease (Rogers, Mastroeni et al. 2007), HIV dementia (Dheen, Kaur et al. 2007) as well as numerous other pathological conditions. Once activated, microglia produce soluble proinflammatory cytokines, prostaglandins and interleukins, such as tumor necrosis factor-α (TNF-α), PGE2, interleukin-1 (IL-1) and interleukin-6 (IL-6), which not only contribute to the neighboring neuronal damage, but also recruit immune cells into the CNS.
The culture technique introduced here is based on the method first published by Barger and Basile (Barger and Basile 2001) and has undergone minor modification in our laboratory. From one litter of 1-day old neonatal rats (10–14 pups) we routinely get sufficient amount of mixed cells (100 millions) for approximately 15 225 cm2 culture flasks. These mixed cell cultures reach confluence in approximately 2 weeks post seeding; the culture media are changed twice weekly. Microglia are isolated from the mixed cell culture before the end of the 2nd week by gentle physical shaking and tapping on the flasks. After isolation, the microglia are re-seeded into three 6-well plates (other seeding options are possible and should be dictated by the end-points of the experiments you wish to conduct taking into account the numbers of replicates) with fresh culture media. The cell purity is routinely examined by immunohistochemistry staining for OX-42, a microglia specific integrin marker. In our hands, the cultures reach >95% purity. Other cells types in the cultures, mainly astrocytes in the mixed culture system lend the process ideal for the concomitant isolation of primary astrocyte cultures. These can be characterized with specific antibodies for intermediate skeletal proteins, such as glial fibrillary acidic protein (GFAP). The method for culturing and maintaining astrocytes has been previously published (Aschner and Allen 2000).
ISOLATION AND MAINTENANCE OF PRIMARY MICROGLIAL CELL CULTURES
MATERIALS
Solutions and Reagents
Coating Cell Culture Plates
70% ethanol
Borate buffer
Borax (71997), Sigma, 3050 Spruce St. St. Louis, MO 63103;
Poly-L-lysine (P-1274), Sigma, 3050 Spruce St. St. Louis, MO 63103;
Autoclaved distilled water
Minimum Essential Medium (MEM) culture media, Gibco, Invitrogen, Grand Island, NY (11095)
Minimum Essential Media, with Earle’s salts, modified for suspension cultures (S-MEM), Gibco, Invitrogen, Grand Island, NY (11385).
Mixed Glial Cell Isolation
70% ethanol
Minimum Essential Medium (MEM) culture media, Gibco, Invitrogen, Grand Island, NY
Dissociation media
Heat inactivated horse serum Gibco, Invitrogen, Grand Island, NY (26050070)
Heat-inactivated fetal bovine serum, Gibco, Invitrogen, Grand Island, NY (10082-147).
Penicillin/Streptomycin, Gibco, Invitrogen, Grand Island, NY (15140-163).
Trypan blue staining solution, (T8154, Sigma)
Hank’s Buffered Salt Solution (HBSS), H2387, Sigma
Sigmacote® (SL-2), Sigma, 3050 Spruce St. St. Louis, MO 63103.
Microglia isolation
70% ethanol
Minimum Essential Medium (MEM) culture media, Gibco, Invitrogen, Grand Island, NY
Dissociation media
Heat inactivated horse serum Gibco, Invitrogen, Grand Island, NY (26050070)
Heat-inactivated fetal bovine serum, Gibco, Invitrogen, Grand Island, NY (10082-147).
Penicillin/Streptomycin, Gibco, Invitrogen, Grand Island, NY (15140-163).
Trypan blue staining solution, (T8154, Sigma)
Maintenance of Cultures
70% ethanol
Heat inactivated horse serum Gibco, Invitrogen, Grand Island, NY (26050070)
Heat-inactivated fetal bovine serum, Gibco, Invitrogen, Grand Island, NY (10082-147).
Penicillin/Streptomycin, Gibco, Invitrogen, Grand Island, NY (15140-163).
Minimum Essential Medium (MEM) culture media, Gibco, Invitrogen, Grand Island, NY
Phosphate buffered saline (PBS) (554781, BD biosciences, San Diego, CA 92121, USA)
Equipment
Coating Cell Culture Plates
Autoclave
6-well cell culture plate
Laminar-flow cell culture hood with ultraviolet (UV) light
Portable power pipette filler/dispenser
Mixed Glial Cell Isolation
Autoclave
Laminar -flow cell culture hood with ultraviolet (UV) light
CO2 incubator (37°C, 95% room air/5% CO2, 95% humidity)
Dissecting microscope
Low speed (60 rpm) stir plate
Low speed centrifuge (<1000 × g) with swinging bucket rotor and 50ml conical tube adapters
Portable power pipette filler/dispenser
Inverted phase-contrast microscope
Hemocytometer
Hand-held counter
Microglial isolation
Autoclave
Laminar -flow cell culture hood with ultraviolet (UV) light
CO2 incubator (37°C, 95% room air/5% CO2, 95% humidity)
Dissecting microscope
Low speed (60 rpm) stir plate
Low speed centrifuge (<1000 × g) with swinging bucket rotor and 50ml conical tube adapters
Portable power pipette filler/dispenser
Inverted phase-contrast microscope
Hemocytometer
Hand-held counter
Maintenance of Cell Cultures
Autoclave
Laminar -flow cell culture hood with ultraviolet (UV) light
CO2 incubator (37°C, 95% room air/5% CO2, 95% humidity)
Portable power pipette filler/dispenser
Disposable Tools for Animal Dissection and Cell Dissociation
Mixed Glial Cell Isolation
Sterile pipettes, disposable borosilicate glass: 146 mm (5 ¾ inch) and 229 mm (9 inch)
Serological pipettes, cotton-plugged disposable borosilicate glass, 10 ml short length
Sterile 5 ml disposable syringes (53500-420, VWR, Batavia, IL 60510, USA)
Syringe filter with 25 cm cellulose acetate membrane, 0.2 μm pore size (4612, Pall corporation, Ann Arbor, MI, 48103, USA)
Sterile serological pipettes, cotton-plugged disposable polystyrene, individually wrapped: 1 ml, 5 ml, 10 ml, and 25 ml (Midwest Scientific, 280 Vance Road, St. Louis, MO, 63088, USA)
Sterile vented cell culture flasks: 225cm2 (353138, BD falcon, Franklin Lake, NJ, 07417, USA)
4 × 3 inch sterile dressing sponges (82004-740, VWR, Batavia, IL, 60510, USA)
15 ml polystyrene centrifuge tubes, sterile (21008-216, VWR, Batavia, IL, 60510, USA)
50 ml polypropylene tubes, sterile (82018-052, VWR, Batavia, IL, 60510, USA)
Microglial isolation
Sterile serological pipettes, cotton-plugged disposable polystyrene, individually wrapped: 1 ml, 5 ml, 10 ml, and 25 ml (Midwest Scientific, 280 Vance Road, St. Louis, MO, 63088, USA)
50 ml polypropylene tubes, sterile (82018-052, VWR, Batavia, IL, 60510, USA)
6 well cell culture plate (353502, BD falcon, Franklin Lake, NJ, 07417, USA)
Maintenance of Microglial Cultures
Sterile serological pipettes, cotton-plugged disposable polystyrene, individually wrapped: 1 ml, 5 ml, 10 ml, and 25 ml (Midwest Scientific, 280 Vance Road, St. Louis, MO, 63088, USA)
Non-disposable Tools for Animal Dissection and Cell Dissociation
Surgical apparatus is non-disposable and must be autoclaved before use. Clean it thoroughly immediately after use.
Dumont forceps (Pattern #5), 110 mm length, tip 0.1 × 0.06 mm, 2 each (25719-066, VWR, Batavia, IL, 60510, USA)
Curves forceps, 4 inch length, full curve, 0.8 mm tip width, 2 each (82027-392, VWR, Batavia, IL, 60510, USA)
Curves forceps, 4 inch length, full curve, 0.4 mm tip width, 2 each (82027-406, VWR, Batavia, IL, 60510, USA)
Micro-dissecting scissors, 4 inch length, 25 mm angled blade, 3 each (89049-674, VWR, Batavia, IL, 60510, USA)
Mayo scissors, 7 inch length, 50 mm curved blade (95039-258, VWR, Batavia, IL, 60510, USA)
50 ml beaker and stir bar (length, 25 mm) Cover top with foil prior to autoclaving
Pipette sterilizing metal boxes: for 5 ¾ inch Pasteur pipettes, 9 inch Pasteur pipettes, and 10 ml glass serological pipettes (20171-042, VWR, Batavia, IL, 60510, USA)
Surgical instrument tray, autoclavable (62687-069, VWR, Batavia, IL, 60510, USA)
BASIC PROTOCOL
The algorithm is listed below:
Time-dated Pregnant Rat Delivery Schedule
We schedule pathogen-free time-pregnant (prenatal day 14; PD14) Sprague-Dawley rats to arrive at the animal facility one week prior to their delivery date (PD21). The number of ordered pregnant rats is calculated based on the fact that one female Sprague-Dawley rat normally gives birth to 10–14 pups. The time-dated pregnant rat delivery allows us to isolate cultures on a consistent basis. We prepare cultures every Wednesday afternoon using 1-day-old pups, so we routinely order pregnant rats for Tuesday delivery to allow them to acclimate for a week prior to delivery. This also is cost effective, as we do not need to maintain our own breeding colony and incur extensive per diem charges. In the long run we find it cost effective as it also frees our staff and students from frequenting the animal facility for husbandry checkups, determination of plugs (PD1), etc.
Mixed Glial Cell Preparation
The protocol described here is composed of two continuous steps. The first step focuses on dissecting rat cortices and the second one entails the dissociation of glial cells including astrocytes, microglia and small amount of oligodendrocytes. These two steps should be performed consecutively.
Step 1: Dissection of Neonatal Rat Cortices
Gently hold and rinse the head and neck of the pup with 70% ethanol.
Decapitate the head on to a sterile gauze pad using sterile curved scissors. The body is placed in the bio-hazard plastic bag for disposal. The surgical apparatus is placed in the 70% ethanol between decapitations to avoid contamination.
Use sterile fine angled dissecting scissors to cut the skin as well as skull at a slight upward angle from the foramen magnum to the eyes along the midline. Extra caution is needed to avoid damaging the brain cortices. The skull flaps can be very easily cut off to simplify the removal of the brain tissue
With the 0.8 mm tip curved forceps sever the olfactory bulbs at the anterior part and the spinal cord at the posterior part of the brain. Gently move forceps with a slight back angle pulling up the brain. This will separate the whole brain from the skull base. During the procedure, the optical nerves can be easily identified; it is not necessary to preserve them.
Place the brain in HBSS in 100mm Petri dish on ice and observe the separated brain under the dissecting microscope.
Gently slip one side of the 0.4 mm tip curved forceps under the corticies on either side of the brain so the forceps sit astride the brain.
Gently move forceps from side-to-side and with a slight back angle pull up of the cortices. This will separate the cortices from the rest of the brain.
With the Dumont forceps, gently tease away the meninges coverings on the cortical surface without damaging it.
Flip over the cortices to expose the underside of the tissue to make sure remove any extra tissues.
You should be able to see the darker hippocampal crescents that need to be gently removed with the curved forceps.
Once each cortex is dissected, place in a sterile 50 ml conical tube containing 10 ml S-MEM + antibiotics on ice. Limit the total procedure within 10 minutes to minimize the damage. We are able to finish the whole procedures within 5 minutes after practice.
Repeat the same procedure in the rest pups.
Place the cortices in the 50 ml tube on ice until the desired number of pup brains has been dissected.
Step 2: Dissociation of Glial Cells
Carefully remove as much of the S-MEM as possible with a sterile cotton-plugged 9” Pasteur pipette taking care to retain all of the cortices.
Add 12 ml of 37°C dissociation media to the 50 ml beaker and gently pulverize the cortices 7–8 times using a Sigmacote® treated (see Support Protocol 1) 10 ml glass pipette.
Stir for 10 minutes at low speed (1 revolution per second) in the laminar-flow hood.
While this is stirring, prepare two 15 ml sterile conical tubes for each extraction. Add 5 ml of growth media to each tube. At this time, also thaw the DNase I and place it on ice.
After ten minutes of gentle stirring, remove the beaker from the stir plate and place at a 45° angle for 2 – 3 minutes to allow the non-dissociated tissue to collect at the bottom of the beaker. Resting the edge of the beaker on a lid from a culture dish works well.
Carefully aspirate 10 ml of the dissociated cells with a Sigmacote® treated 10 ml glass pipette. Take care not to disturb the undissociated tissue pieces.
Place 5 ml into each of the two 15 ml centrifuge tubes containing the 5 ml of growth media. Invert the mixture 2 – 3 times to mix the cell suspension and growth media. The serum in the growth media acts to inhibit dispase and prevents over digestion of the dissociated cells.
Add another 10 ml of 37°C dissociation media to the 50 ml beaker and add 100 μl of DNase I. Continue to stir for another 10 minutes. Note the DNase I is added only after the first extraction and is not added again.
Place the 50 ml beaker at an angle for 2–3 minutes, remove 10 ml of dissociated cells, and place 5 ml into another pair of 15 ml tubes containing 5 ml growth media as before.
Add another 10 ml of 37°C dissociation media to the tissue in the 50 ml beaker and stir for 10 minutes. (Do not add DNase I again.) The removal of dissociated cells and replacement with 10 ml dissociation media is called an extraction.
Extractions are repeated until there is only fibrous tissue remaining in the 50 ml beaker. The number of extractions is usually equal to the number of dissected brains.
Allow each pair of 15 ml conical tubes to sit undisturbed during the continuing extractions. During this time, undissociated tissue will settle to the bottom of the 15 ml tube. This undissociated tissue is placed back into the 50 ml beaker for further dissociation during the final two extractions.
To remove undissociated tissue from the 15 ml tubes, insert a sterile cotton-plugged Sigmacote® treated 9” Pasteur pipette to the bottom of the tube and carefully aspirate the undissociated tissue. Place this tissue back into the 50 ml beaker for further dissociation. This is done only during the final two extractions because serum carried over from the completed extractions can inactivate the dispase.
Once all of the extractions have been processed, pool the dissociated cells and media from the 15 ml centrifuge tubes into 50 ml conical tubes. Centrifuge the 50 ml tubes for 10 minutes at 500 rpm in a swinging bucket rotor to pellet the suspended cells.
Carefully aspirate the media from the cell pellets.
Resuspend the cells in 20 ml growth media per 50 ml tube by gentle pipetting with a 10 ml Sigmacote® treated glass pipette.
Allow cells to sit for an additional 5 minutes and remove any sedimented tissue as before with a 9” Pasteur pipette. Discard this undissociated tissue.
Pool the suspended cells into 50 ml tubes on ice.
Determine the total and viable cells by gently mixing 100 μl cells with 100 μl diluted trypan blue staining solution. Allow the cells to take up the trypan blue for 5 min and determine cell number and viability with a hemocytometer.
Dilute cell suspension to 10,000 viable cells/ml with growth media.
Pipette 2,500 viable cells/cm2 into 225 cm2 culture plates and then add growth media to the final volume of 75 ml.
Unlike culturing astrocytes, the media must not be changed until 7 days after plating to allow mixed glial cells to attach. Approximately 5~ 10 % of the mixed glial cells plated will attach and grow on the coated plastic surface over the next 1.5~ 2.5 weeks.
When changing media, insert the 5 inch Pasteur pipette and aspirate the media. Change the Pasteur pipette frequently and when it may have touched anything but inside of the culture flasks. A pipette contaminated during media changes can easily infect the entire culture if it is not changed.
Add 75ml fresh culture media to using a large disposable pipette.
Media is changed twice per week. If the culture media turn turbid, the cells should be discarded and the possibility of contamination should be suspected.
Isolation of Microglia from the Mixed Glial Culture
Check the mixed glial cell culture under inverted phase-contrast microscope approximately 2 weeks after the initial plating, when astrocytes reach 100% confluence. The microglia are growing on the top of single layer of astrocytes as small rounded cells. Adjusting the fine focus of the microscope assists in distinguishing the different cell layers.
Put the cell culture flask on the flatten surface and gently bang on the side and tap the flasks at the speed of 45 times per minutes. Try to minimize the amount of foam generated when shaking the cell culture flasks.
While tapping and shaking the cell culture flask, prepare the centrifuge. In our laboratory, we use a PRECISION Durafuge 200R centrifuge (PRECISION, 170 Marcel Drive, Winchester, VA, USA) and set the temperature to 4°C. Based on our experience, it takes about 20 minutes to cool down from room temperature to 4°C. This may vary based on the type of centrifuge available to you so make sure it is ready for use at the appropriate temperature as you are isolating the cells to prevent any delays.
Check the cell culture flask under the inverted phase- contrast microscope every 3 minutes to make sure the monolayer astrocytes are not disturbed and continue to adhere to the substratum of the flask.
The detached microglia float in the media, while the astrocytes remain attached to the bottom.
If attached microglia are observed, keep shaking the flasks until almost all microglia are floating. Using this method, after gentle shaking of the flasks for 9–12 minutes we are able to get almost all of the microglia without disturbing the adherent astrocytes.
If astrocytes are sheared from the bottom, immediately stop shaking the flasks. If the harvested cells are contaminated with significant amounts of astrocytes, this may be caused by two reasons: 1) shaking is too rigorous; 2) astrocytes are not sufficiently healthy to remain attached to the substratum of the flask. If astrocytes readily detach from the flask consider changing fresh culture media more frequently (three rather than two times per week). Please refer to the Troubleshooting section for additional details.
After most microglia detach, stop shaking and carefully aspirate the culture media and put it into pre-chilled 50 ml conical tube.
Spin down the cells at 1000 RPM at 4°C for 5 minutes using PRECISION Durafuge 200R (or a similar device) with a swinging bucket rotor to pellet the suspended cells.
Carefully aspirate the media without disturbing the cell pellets.
Resuspend the cells in 5 ml growth media per 50 ml tube by gentle pipetting with a 10 ml Sigmacote® treated glass pipette.
Pool the suspended cells into 50 ml tubes on ice.
Determine the total and viable cells by gently mixing 100 μl of cells with 100 μl of diluted trypan blue staining solution. Allow the cells to take up the trypan blue for 5 minutes and determine cell number and viability with a hemocytometer. Stained cells are dead; those that exclude the trypan blue are considered alive.
Dilute cell suspension to 20,000 viable cells/ml with growth media.
Pipette 5,000 viable cells/cm2 into each well of six-well cell culture plates and add growth media to a final volume of 2ml/well.
The isolated primary microglia are maintained in a CO2 incubator (NAPCO series 8000DH, Thermo Scientific, San Diego, CA 92121, USA) (37°C, 95% room air/5% CO2, 95% humidity) for 48 hours before the experiments are initiated. Our experience dictates the purity of isolated microglia culture is >95% [immunostaining for the microglial specific marker, OX42 (Fig 1)].
The leftover cells in the culture flasks are mainly astrocytes and they are removed by trypsinization. The protocol introduced here is ideal for the concomitant isolation of both microglia and astrocytes. The detailed protocol of isolation and culturing primary astrocytes has been published by our laboratory before (Yin, Aschner et al. 2008). Trypsin is most effective at a pH between 7.0 and 8.0 and between 24°C and 37°C at 0.05 % to 0.5 % concentration. The trypsinization time required for cells removal from the plates depends on their density, serum concentration and temperature. EDTA is often added to the trypsin to intensify enzyme activity by chelating calcium and magnesium.
Add 10 ml of warm phosphate buffered saline (PBS) and rock the flask to wash he monolayer briefly. This assures the removal of the last traces of serum. Astrocytes should not be left in PBS for more than 10 minutes to avoid excessive cell damage.
Aspirate the PBS with a Pasteur pipette.
Add 2ml 1X trypsin (37°C) and rock the flask gently to cover the entire surface with trypsin.
Leave the flask in a CO2 incubator (37°C, 95% room air/5% CO2, 95% humidity) for 5 minutes.
When the cells are loose, add 10 ml of fresh culture media with serum and resuspend the cells.
Dilute cells to 10,000 viable cells/ml growth media.
Pipet 2,500 viable cells/cm2 into each well of the six-well cell culture plates and add growth media to a final volume of 2ml/well. The isolated astrocytes are maintained at CO2 incubator (37°C, 95% room air/5% CO2, 95% humidity) for 5 minutes. The purity of astrocytes in our hands reaches >95%. We routinely test the purity by immunohistochemistry staining for the astrocyte specific marker, GFAP (Fig 2).
Figure 1.

The purity of isolated primary microglia exceeds 95% as verified by immunohistochemistry staining for the microglia specific marker, OX 42. The nuclei are counterstained with 4′,6- diamidino- 2- phenylindole (DAPI).
Figure 2.
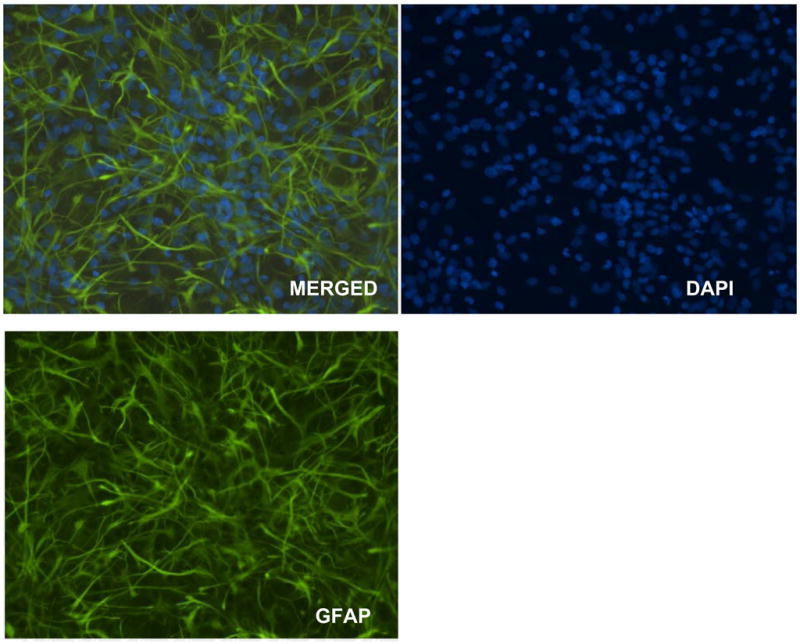
The purity of leftover astrocytes exceeds 95% as verified by immunohistochemistry staining for the astrocyte specific marker, GFAP. The nuclei are counterstained with DAPI.
Support Protocols
Support Protocol 1: Fire Polishing and Sigmacoat® Treatment of Pipettes for Cell Isolation
The fire polished and Sigmacote® treated pipettes are only used for cell isolation, not routine changing of media. These pipettes need to be fire polished and Sigmacoat® treated. In our laboratory, we routinely fire a batch of 50 pipettes for each planned microglial cell isolation.
Fire polished tips of 9 inch Pasteur pipettes by placing the small end in an open flame from a Bunsen burner for a few seconds. This will slightly melt the borosilicate glass and will produce a smooth tip to the pipette which helps prevent damage to the cells. Take care not to make the diameter of the opening too small as this can increase the shear forces during triturating and may lead to increased cell damage.
Plug the large end of the Pasteur pipettes with cotton. The amount of cotton obtained from a single cotton tipped swab is sufficient for each pipette.
Treat each pipette with Sigmacote® by drawing the viscous solution up into the Pasteur pipette without touching the cotton plug. Allow the Sigmacote® to drain back into the bottle and place the Pasteur pipette in a beaker with paper towels lining the bottom to drain and dry completely.
Once the Pasteur pipettes are dried, place them in a metal container and autoclave.10 ml glass serological pipettes should also be Sigmacote® treated and autoclaved. Fire polishing of these pipettes is unnecessary.
Supporting Protocol 2: Coating Cell Culture Plate with Poly-L-Lysine
We routinely culture isolated microglia in the poly-L-lysine coated six-well cell culture plate. Prepare borate buffer according the following recipe:
prepare coating buffer by dissolving 2mg poly-L-lysine into 50ml borate buffer;
add 1 ml coating buffer into each well;
let it stand for 1 hour in the tissue culture hood with UV light on;
aspirate the coating buffer and wash the well with distilled water for three times
air dry the cell culture plate in the tissue culture hood for at least one hour with UV light on;
Add 1 ml MEM culture media without antibiotics or serum into each well and store the plate in 4°C. The coated plate could be stored safely for one week.
REAGENTS AND SOLUTIONS
Reagents
-
Culture Media:
Minimal Essential Medium, with Earle’s salts and L-Glutamate (MEM), Gibco, Invitrogen, Grand Island, NY (11095)
Minimal Essential Media, with Earle’s salts, modified for suspension cultures (S-MEM), Gibco, Invitrogen, Grand Island, NY (11385). S-MEM has been modified to contain no Ca+2 which can produce cell clumping due to interactions of extracellular matrix proteins.
-
Antibiotics:
Penicillin G (10,000U/ml)-Streptomycin Sulfate (10,000 μg/ml), Gibco, Invitrogen, Grand Island, NY (15140-163). Avoid repeated freeze- thaw cycles of penicillin/streptomycin by storing 10ml aliquots at −20°C. 10 ml penicillin/streptomycin is enough for 1 L of media.
Fungizone® (Amphotericin B, 250 μg/ml), Gibco, Invitrogen, Grand Island, NY (15290-018). Fungizone is optional, to prevent fungal or yeast contamination.
-
Serum:
Heat-inactivated horse serum, Gibco, Invitrogen, Grand Island, NY (26050070)
Heat-inactivated fetal bovine serum, Gibco, Invitrogen, Grand Island, NY (10082-147). Avoid repeated freeze-thaw cycles of serum. Put the frozen serum in the water bath no warmer than 40°C until serum is thawed. The thawed serum could be safely stored at 4°C for approximately one week.
Protease (dispase), Gibco, Invitrogen, Grand Island, NY (17105-041).
Deoxyribonuclease I (DNase I) from Bovine Pancreas Type IV, Sigma Chemical Company, St. Louis, MO (D-5025). DNase I is added immediately after the first extraction to prevent the genomic DNA released by damaged cells from making the dissociating media too viscous during the ongoing digestion. Avoid vortex when handling DNase I as it is vulnerable to inactivation by physical damage.
Poly-L-lysine Hydrobromide (P-1274), Sigma, 3050 Spruce St. St. Louis, MO 63103.
Trypan blue 0.4%, liquid, sterile-filtered, cell culture tested (T8154). Trypan blue is used determine cell viability. Dead or damaged cells cannot exclude trypan blue, so they appear blue under the microscope.
Sigmacote® (SL-2), Sigma, 3050 Spruce St. St. Louis, MO 63103. Sigmacote is a special silicone solution in heptane that readily forms a covalent, microscopically thin film on glass, retards clotting of blood or plasma. It bonds with the uneven surface of the glass pipettes and provides a smooth hydrophobic barrier. The adherence of cells to the glass surface is greatly reduced, and thus minimizing physical damage to the cells.
Distilled Water, cell culture tested, Gibco, Invitrogen, Grand Island, NY (15230-147). All solutions used in this protocol should be prepared in cell culture tested double distilled water.
Boric Acid, J.T. Baker, 222 Red School Lane, Phillipsburg, NJ (10043-35-3).
Borax (71997), Sigma, 3050 Spruce St. St. Louis, MO 63103.
CRITICAL PARAMETERS
Dissecting neonatal rat cortex
The meninges should be completely removed; otherwise the mixed glial culture may become contaminated with large amount of fibroblasts as the proliferation of fibroblast is much faster than glial cells. An efficient surgical technique is critical for obtaining high quality tissue dissection. Avoid spending more than 10 minutes on dissecting brains to minimize the ischemic damage to the brain tissue.
Dissociation of cells
According to our experience, there is significant variability in the quality of dispase used in this protocol. In general, if a rapid and continuing decrease in cell number or viability is observed, dispase should be changed to newly purchased lots. The use of Sigmacote® treated glass pipettes greatly enhances the quality of dissociated cells because it significantly reduces the trauma to cells during the isolation process. We have observed that harsh maneuver results in a large reduction in the cell viability.
Isolation of Primary Microglia
In general, isolation of microglia by shaking flasks should be gentle to avoid forceful trituration that introduces air bubbles into the suspensions. It is important to change culture media twice a week (or more, should astrocytes detach from the substratum during the tapping process) to keep cells healthy; otherwise, astrocytes could be sheared during the isolation procedure very easily. Keep checking the cells under the inverted microscope every 3 minutes in order to minimize the amount of astrocyte contamination.
Cell Culture Purity
The primary microglial culture obtained by our protocol is routinely >95% positive for OX 42 (Castellano, Gonzalez et al. 1991) by immunohistochemistry (See Figure 1). Unlike non-glial cells, microglia should be cultured in Poly-L-lysine or collagen I coated plates. It has been suggested that using rat pups 24 to 48 hours old may decrease the number of contaminated neurons (McCarthy and de Vellis 1980). The small spikes in primary microglia could be very easily observed under microscope with DIC settings (Fig 1).
Coating Material
Primary microglia should not be cultured on uncoated plastic surface or glass surface. In our laboratory, we used both poly-L-Lysine and collagen I coated cell culture plates. One of the unique features of primary microglia is that the phenotype could be different based on different coating materials. Cells are wide spread on Poly-L-lysine coated surface, but microglia grow in clusters on the collagen I coated surface. Therefore, choosing which coating material to use should be carefully considered within the context of the experimental design.
Culture Time
Unlike malignant cells, primary microglia stop proliferating and growing once confluence is reached. Consideration should also be directed at the possibility that different confluence level may cause significant variations in experiment results (Garg and Chang 2006). Therefore, it is strongly recommended that the effects of confluence be determined by treating cultured microglia at different confluence levels. Furthermore, experiments should be conducted in triplicates (at a minimum) derived from several independently isolated cultures. Experiments must use cultured cells at the same confluence in order to minimize this confounding factor. At our hands cultured microglia reach 50% confluence two weeks after isolation from the mixed glial culture preparations.
Contamination
If the culture media turn turbid, contamination should be suspected and all infected culture flasks must be disposed immediately. Rapid and accurate identification of the offending microbe is very helpful, but rarely could be done in the laboratory settings. The most common pathogenic microbial include Staphylococcus aureus, Staphylococcus epidermidis or Streptococci, though some rare microbe could be possible such as microplasma, fungi or viruses. The contamination caused by microplasma or viruses could be very difficult to identify because the culture media do not turn turbid. The only sign of microplasma or virus infection is that a large number of cells fail to attach to the substratum. The microbial contamination is most likely to occur during the initial cell isolation, thus sterilization of surgical tools and cleaning of all surface with 70% ethanol are critical to minimize microbial contamination.
TROUBLESHOOTING
Clumping of cells during the extraction
Cell clumping causes significant cell damage during the cell isolation. Ca2+ in the regular MEM could interact with the extracellular proteins, which lead to cell clumping. The cell clumping should be differentiated from tissue sedimentation by its size. Cell clumping is much smaller than tissue sedimentation and checking the sedimentation under microscope could be very easily tell them apart. It is recommended to use S-MEM without Ca2+ during cell isolation to prevent cell clumping.
Media becomes very viscous during extraction
It is caused by genomic DNA release from damaged cells and adding DNase I immediately after the first extraction could very well solve it. The DNase I solution should not be vortexed as it is vulnerable to inactivation by physical damage.
Large area of monolayer of astrocytes sheared off during isolation of primary microglia
It is most likely caused by too vigorous shaking of the cell culture flask, especially if the researcher has limited experience in the isolation procedure. We recommend starting with less vigorous physical maneuver and checking the cells under the inverted microscope every three minutes, adjusting the level of vigorousness based on the isolation outcome. Changing the old with fresh culture media could also help to mitigate this problem.
COMMENTARY
Microglia play a critical role as resident immunocompetent and phagocytic cells, serving as scavenger cells in the event of infection, inflammation, trauma, ischemia and neurodegeneration. Thanks to the advancement of cell culture techniques, research on the biochemical, physiological, pharmacological and molecular aspects of microglial function are possible. The culture technique provides an ideal model to study the microglial function under controlled and reproducible conditions.
Given the wide array of microglial functions, the possible uses for primary cultures are endless. A brief list includes, studies on the role of microglia in immunity (Yang, Han et al. 2009), protective effects of microglia on co-cultured neurons (Park, Zhang et al. 2001), modulators of microglial secretion of growth factors (Wang, Cui et al. 2009) and cytokines (Ye and Johnson 1999; Chang 2007), and the role of free-radicals production in neuropathology (Long, Tajuba et al. 2007), just to name a few. The receptors of microglia contribute to the modulation of CNS microenvironment and functions. For example, the main functional ionotropic glutamate receptors (iGluRs) in cultured rat microglia are mostly AMPA (D,L-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor, which inhibits TNF-α release and helps to modulate glutamate level in the CNS. Furthermore, perturbations in the homeostasis of this transmitter have been reported in neurodegenerative diseases (Pocock and Kettenmann 2007). Microglia also express GABAB (γ-aminobutyric acid) receptor that inhibits IL-6 release from the activated microglia (Pocock and Kettenmann 2007). In addition, these cells express a variety of purinergic receptors (P2Y12, P2Y6, P2Y7 AND P2Y4), which regulate the migration and cytokine release. Furthermore, microglia exhibit anti-inflammatory effects by expressing adrenergic, dopaminergic and cholinergic receptors (Pocock and Kettenmann 2007). Studies on the function of the above parameters are all amenable to in vitro cultured microglia, and indeed, over the last several decades a plethora of experimental approaches have enriched our understanding on the role of these cells in health and disease.
Though microglial cells can be readily isolated and have clearly received experimental attention, many studies have utilized microglial cell lines instead of primary cells to test their functions in various physiological and pathological conditions(Garg and Chang 2006). Although immortalized cell lines, such as murine N9 are less expensive and less time consuming to prepare and maintain, and there is less culture- to- culture variation in these cell lines, it needs to be considered that cell lines are derived from either tumors or are genetically modified to proliferate unlimitedly. Thus results generated from cell lines may lead to erroneous interpretations. Primary microglia have advantages over the immortalized cells more closely representing their in situ counterparts. However, analogous to any type of cultured cells, one must be cautious when relating processes seen in cultured microglia and extrapolating to in vivo conditions as primary cells are grown in isolation and lack the normal CNS microenvironment. The interaction between multiple cell types in the brain such as neurons, astrocytes, oligodendrocytes and microglia is exceedingly difficult to be modeled in vitro. Regardless what isolation protocol is used (for additional methods see below), it is important to keep in mind that one must be cautious in correlating the in vitro data generated from cultured primary microglia with their physiological and pathological profile in living animals. Issues related to the advantages and disadvantages offered by cell cultures have been previously addressed and apply to any cell type.
There are additional published methods to isolate primary microglia. Frank and Maier (Frank, Wieseler-Frank et al. 2006) reported a density gradient centrifugation method of homogenized hippocampal tissue in a 0/50/70% Percoll (Amersham Biosciences, Uppsala, Sweden) gradient that yields enriched rat primary microglia. The primary microglial cells are located at the 50/70% Percoll interface. The disadvantages of this protocol include long centrifugation (45 minutes) at 20°C as well as the homogenization of brain tissue before the spin down. The homogenization procedure may cause excessive cell damage and the subsequent long spin down time may further decrease the microglial yield, thus selecting for a specific (potentially hardy) microglial phenotype. Hassan and Douglas introduced another protocol to isolate human (Hassan, Campbell et al. 1991), mouse (Hassan, Rifat et al. 1991) and rabbit (Hassan, Prakash et al. 1991) primary microglia using orbital shaker. Briefly, flasks with the mixed glial culture are shaken in an orbital shaker for 16 h at 150 rpm at 37°C. The supernatant media are then centrifuged for 10 minutes at 1,200 r.p.m at 4°C. This method is short and saves labor, and also yields large amounts of primary microglia. Nevertheless, the sequalae of 16 h shaking on the functional and morphological properties of microglia needs to be considered and evaluated.
We do not recommend passaging of the mixed glial cell cultures. One may consider trypsinizing mixed glial cultures, splitting cells into more culture flasks and allowing them to grow until confluence (passage of cells). Although this will result in a greater glial cell yields, it should be considered that cell passaging may change their biochemical and immunological phenotypes (Cole and de Vellis, 1992). Therefore, we do not recommend this approach and advocate fresh isolations of microglia to obtain a constant supply of primary cells. .
Regardless what isolation protocol used, it is important to keep in mind that one must be cautious to correlate the in vitro data generated from cultured primary microglia with the physiological and pathological profile in the living animals.
CELLULAR YIELDS
The protocol described above results in the isolation of microglial cultures that exceed 95% purity [by immunohistochemical staining for the specific marker, OX42 (Fig 1)]. The primary microglia seeded and grown on Poly-L-lysine coated plastic surface do not grow in clusters, so that fine structures on the cell membrane could be observed in detail (Fig 3A). Conversely, cultured primary microglia seeded on Collagen I grow in clusters (Fig 3B). Cell yields reach 1,000,000 microglial cells per brain with viability approximating 90%. Most isolated primary microglia (>90%) adhere to the coated cell culture plates 24 hours after the isolation. The fresh culture media can be changed 72 hours after initial isolation if necessary. Our experience dictates that 6,000,000–8,000,000 cultured primary microglia provide ample RNA or protein for most real-time PCR or western blot procedure.
Figure 3.

DIC images of cultures primary microglial cells. Fig. 3A shows the isolated primary microglial cells cultured on Poly-L-lysine coated plastic surface and Fig. 3B shows the cells cultured on Collagen I coated surface. Different coating materials produce different morphological attributes in these cells. In general, cells are widely spread when grown on poly-L-lysine (Fig. 3A) coated plastic surface with small spikes easily observed. Collagen I causes the microglia to grow in clusters (Fig. 3B).
TIME CONSIDERATIONS
The total time spent on the preparation of mixed glial culture is approximately 6 hours depending on the number of extractions performed and the number of rat pups used. The mixed glial cell culture is maintained in 225 cm2 cell culture flasks on average for 2 weeks (dependent upon the endpoint of your experiment; obviously if you desire to study developmental changes, you may study the cells at earlier time points, keeping in mind that replicates should be at the same developmental stage and confluence). The twice weekly changing of media takes about 2 hours based on the number of flasks. It is recommended to feed the cells more frequently if the astrocytes adhere to the bottom very loosely. The isolation of primary microglia usually takes about 3 hours.
Acknowledgments
This chapter was supported in part by funds from the National Institute of Environmental Health Sciences, NIEHS 10563 and 07731 (MA).
Contributor Information
Mingwei Ni, Email: Mingwei.ni@vanderbilt.edu.
Michael Aschner, Email: Michael.aschner@vanderbilt.edu.
CITATION
- Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15(5):542–8. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Aschner M, Allen JW. Astrocytes in methylmercury, ammonia, methionine sulfoximine and alcohol-induced neurotoxicity. Neurotoxicology. 2000;21(4):573–9. [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76(3):846–54. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Castellano B, Gonzalez B, et al. A double staining technique for simultaneous demonstration of astrocytes and microglia in brain sections and astroglial cell cultures. J Histochem Cytochem. 1991;39(5):561–8. doi: 10.1177/39.5.1707903. [DOI] [PubMed] [Google Scholar]
- Chang JY. Methylmercury causes glial IL-6 release. Neurosci Lett. 2007;416(3):217–20. doi: 10.1016/j.neulet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- Cole R, de Vellis J. Astrocyte and oligodendrocyte cultures. In: Federoff S, Richardson A, editors. Protocols for Neural Cell Culture. Humana; Totowa, NJ: 1992. pp. 65–80. [Google Scholar]
- Dheen ST, Kaur C, et al. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14(11):1189–97. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, et al. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods. 2006;151(2):121–30. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. J Neuroimmunol. 2006;171(1–2):17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Hamilton JA, Hillard CJ, et al. Brain uptake and utilization of fatty acids, lipids and lipoproteins: application to neurological disorders. J Mol Neurosci. 2007;33(1):2–11. doi: 10.1007/s12031-007-0060-1. [DOI] [PubMed] [Google Scholar]
- Hassan NF, Campbell DE, et al. Isolation and characterization of human fetal brain-derived microglia in in vitro culture. Neuroscience. 1991;41(1):149–58. doi: 10.1016/0306-4522(91)90205-3. [DOI] [PubMed] [Google Scholar]
- Hassan NF, Prakash K, et al. Isolation and characterization of newborn rabbit brain-derived microglia. Clin Immunol Immunopathol. 1991;59(3):426–35. doi: 10.1016/0090-1229(91)90038-c. [DOI] [PubMed] [Google Scholar]
- Hassan NF, Rifat S, et al. Isolation and flow cytometric characterization of newborn mouse brain-derived microglia maintained in vitro. J Leukoc Biol. 1991;50(1):86–92. doi: 10.1002/jlb.50.1.86. [DOI] [PubMed] [Google Scholar]
- Long TC, Tajuba J, et al. Nanosize titanium dioxide stimulates reactive oxygen species in brain microglia and damages neurons in vitro. Environ Health Perspect. 2007;115(11):1631–7. doi: 10.1289/ehp.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park LC, Zhang H, et al. Co-culture with astrocytes or microglia protects metabolically impaired neurons. Mech Ageing Dev. 2001;123(1):21–7. doi: 10.1016/s0047-6374(01)00336-0. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30(10):527–35. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, V, Perry H. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rogers J, Mastroeni D, et al. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol. 2007;82:235–46. doi: 10.1016/S0074-7742(07)82012-5. [DOI] [PubMed] [Google Scholar]
- Schwabe T, Bainton RJ, et al. GPCR signaling is required for blood-brain barrier formation in drosophila. Cell. 2005;123(1):133–44. doi: 10.1016/j.cell.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Shie FS, Nivison M, et al. Modulation of microglial innate immunity in Alzheimer’s disease by activation of peroxisome proliferator-activated receptor gamma. Curr Med Chem. 2009;16(6):643–51. doi: 10.2174/092986709787458399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernadakis A. Neuron-glia interrelations. Int Rev Neurobiol. 1988;30:149–224. [PubMed] [Google Scholar]
- Wang Y, Cui X, et al. A critical role of activin A in maturation of mouse peritoneal macrophages in vitro and in vivo. Cell Mol Immunol. 2009;6(5):387–92. doi: 10.1038/cmi.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang I, Han SJ, et al. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci. 2009 doi: 10.1016/j.jocn.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93(1–2):139–48. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- Yin Z, Aschner JL, et al. Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Res. 2008;1203:1–11. doi: 10.1016/j.brainres.2008.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]