

Abstract
Single crystals of Co3(PO4)2·4H2O, tricobalt(II) bis[orthophosphate(V)] tetrahydrate, were obtained under hydrothermal conditions. The title compound is isotypic with its zinc analogue Zn3(PO4)2·4H2O (mineral name hopeite) and contains two independent Co2+ cations. One Co2+ cation exhibits a slightly distorted tetrahedral coordination, while the second, located on a mirror plane, has a distorted octahedral coordination environment. The tetrahedrally coordinated Co2+ is bonded to four O atoms of four PO4 3− anions, whereas the six-coordinate Co2+ is cis-bonded to two phosphate groups and to four O atoms of four water molecules (two of which are located on mirror planes), forming a framework structure. In addition, hydrogen bonds of the type O—H⋯O are present throughout the crystal structure.
Related literature
Besides crystals of the title compound, crystals of Co3(PO4)2·H2O (Lee et al., 2008 ▶) have also been obtained under hydrothermal conditions. For reviews, synthesis, structures and applications of open framework structures with different cations and/or structure directing molecules, see: Kuzicki et al. (2001 ▶); Chen et al. (2006 ▶); Jiang & Gao (2007 ▶); Cheetham et al. (1999 ▶); Forster et al. (2003 ▶); Jiang et al. (2001 ▶); Cooper et al. (2004 ▶); Choudhury et al. (2000 ▶). The structure of the isotypic mineral hopeite was first described by Liebau (1965 ▶).
Experimental
Crystal data
Co3(PO4)3·4H2O
M r = 438.79
Orthorhombic,

a = 10.604 (3) Å
b = 18.288 (5) Å
c = 5.0070 (13) Å
V = 971.0 (5) Å3
Z = 4
Mo Kα radiation
μ = 5.46 mm−1
T = 150 (2) K
0.52 × 0.39 × 0.38 mm
Data collection
Siemens SMART 1000 CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1999 ▶) T min = 0.068, T max = 0.125
8780 measured reflections
1228 independent reflections
1166 reflections with I > 2σ(I)
R int = 0.026
Refinement
R[F 2 > 2σ(F 2)] = 0.035
wR(F 2) = 0.102
S = 1.16
1228 reflections
101 parameters
10 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.55 e Å−3
Δρmin = −1.38 e Å−3
Data collection: SMART (Siemens, 1995 ▶); cell refinement: SAINT (Siemens, 1995 ▶); data reduction: SAINT and XPREP (Siemens, 1995 ▶); program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶), WebLab ViewerPro (Molecular Simulations, 2000 ▶) and POV-RAY (Cason, 2002 ▶); software used to prepare material for publication: enCIFer (Allen et al., 2004 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808028377/wm2193sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808028377/wm2193Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected bond lengths (Å).
| Co1—O7i | 1.901 (3) |
| Co1—O1 | 1.918 (3) |
| Co1—O2ii | 1.983 (3) |
| Co1—O2iii | 1.986 (3) |
| Co2—O4 | 2.106 (4) |
| Co2—O5 | 2.118 (4) |
| Co2—O6 | 2.138 (3) |
| P1—O7 | 1.519 (3) |
| P1—O3 | 1.521 (3) |
| P1—O1 | 1.537 (3) |
| P1—O2 | 1.570 (3) |
Symmetry codes: (i) 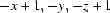 ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O4—H5⋯O3iv | 0.894 (10) | 2.25 (8) | 2.764 (4) | 116 (7) |
| O4—H5⋯O7v | 0.894 (10) | 2.49 (8) | 3.209 (3) | 138 (9) |
| O4—H6⋯O3vi | 0.894 (10) | 2.02 (7) | 2.764 (4) | 140 (10) |
| O5—H4⋯O3vii | 0.892 (10) | 2.25 (2) | 3.133 (5) | 170 (8) |
| O5—H4⋯O3viii | 0.892 (10) | 2.66 (8) | 3.133 (5) | 115 (6) |
| O6—H1⋯O1vii | 0.90 (4) | 1.94 (3) | 2.690 (4) | 140 (4) |
| O6—H2⋯O7 | 0.90 (3) | 2.35 (3) | 3.008 (5) | 130 (3) |
Symmetry codes: (iv)  ; (v)
; (v)  ; (vi)
; (vi)  ; (vii)
; (vii)  ; (viii)
; (viii)  .
.
Acknowledgments
We gratefully acknowledge the Brain Korea 21 programme and the Australian Research Council for support.
supplementary crystallographic information
Comment
The synthesis and investigation of open-framework transition-metal phosphates has been a growing area of research over recent times. This is not only because of the rich structural chemistry involved, but is also due to many potential applications such as for catalysis, as alternatives for zeolites in separation and storage applications and, in particular, as potential gas storage materials (Kuzicki et al., 2001; Chen et al., 2006; Jiang & Gao, 2007; Cheetham et al., 1999). For example, microporous nickel phosphates incorporating 24-membered rings such as VSB-5 (Versailles/Santa Barbara-5) have been demonstrated to exhibit hydrogen uptake at low temperatures (Forster et al., 2003). Over the past couple of decades, a considerable number of metal phosphates/phosphites with open molecular architectures have also been synthesized incorporating organic units (see, for example: Jiang et al., 2001) and ionic liquids (Cooper et al., 2004) as structure-directing agents, often under hydrothermal or solvothermal conditions. One of the best known families of this type consists of zinc phosphate structures; individual materials of this type can exist as one dimensional (chain and ladder), two dimensional (layer) and three dimensional framework arrangements (Choudhury et al., 2000).
We are currently investigating the synthesis of a variety of similar functional materials through templation effects under hydrothermal conditions. The title compound, Co3(PO4)2.4H2O, (I), and the related compound Co3(PO4)2.H2O (Lee et al., 2008) were synthesized and structurally characterized as a part of these studies.
The structure of (I) is isotypic with the mineral hopeite, Zn3(PO4)2.4H2O (Liebau, 1965) and contains two different Co2+ centres bridged by orthophosphate anions (Fig. 1). The coordination environment of Co1 is slightly distorted tetrahedral while that of Co2 is close to octahedral (Table 1). Co1 is bonded to the O atoms of four different phosphate ligands, while Co2 is bonded to the O atoms of two orthophosphate ligands in a cis-arrangement. The other coordination sites are occupied by O atoms of the water ligands. A mirror plane passes through Co2 and two of the water molecules (O4 and O5). This coordination geometry leads to the formation of a three-dimensional framework (Fig. 2). A number of hydrogen bonding interactions O—H···O are present and stabilize the structure (Table 2).
Experimental
H3PO4 (85%wt, 2.3 g, 20 mmol) was added to an aqueous solution (20 ml) of Co(NO3)2.6H2O (2.6 g, 9 mmol) with stirring for 30 min and the ionic liquid, 1-butyl-3-methylimidazolium bromide (2.7 g, 9 mmol), was added dropwise under continuous stirring. The mixture was transferred to a teflon-coated autoclave, heated at 453 K for 3 d and then allowed to cool slowly. A mixture of plate-like and prismatic purple crystals had formed and was filtered off. The crystals were washed with water, dried under vacuum and were manually separated under a microscope. The yields were approximately 0.4 g of the plate-like crystals of the compound Co3(PO4)2.H2O (Lee et al., 2008) and and 0.2 g of the prismatic crystals of compound (I).
Refinement
Water H atoms were located in difference Fourier maps and were refined with Uiso(H) values fixed at 1.5Ueq of the parent O atoms. O—H bond length restraints of 0.89 (1) Å were also employed. The highest peak and the deepest hole in the final Fourier map are located 0.49 Å from Co1 and 0.33 Å from P2, respectively.
Figures
Fig. 1.

The asymmetric unit of compound (I), drawn with displacement parameters at the 50% probability level. H atoms are given as spheres of arbitrary radius.
Fig. 2.

A schematic representation of a section of the three-dimensional network in a projection along [001]. Hydrogen atoms are omitted for clarity.
Crystal data
| Co3(PO4)3·4H2O | F(000) = 860 |
| Mr = 438.79 | Dx = 3.002 Mg m−3 |
| Orthorhombic, Pnma | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ac 2n | Cell parameters from 5943 reflections |
| a = 10.604 (3) Å | θ = 2.9–28.3° |
| b = 18.288 (5) Å | µ = 5.46 mm−1 |
| c = 5.0070 (13) Å | T = 150 K |
| V = 971.0 (5) Å3 | Prism, purple |
| Z = 4 | 0.52 × 0.39 × 0.38 mm |
Data collection
| Siemens SMART 1000 CCD diffractometer | 1228 independent reflections |
| Radiation source: sealed tube | 1166 reflections with I > 2σ(I) |
| graphite | Rint = 0.026 |
| ω scans | θmax = 28.3°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1999) | h = −13→13 |
| Tmin = 0.068, Tmax = 0.125 | k = −24→24 |
| 8780 measured reflections | l = −6→6 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.102 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.16 | w = 1/[σ2(Fo2) + (0.059P)2 + 3.8213P] where P = (Fo2 + 2Fc2)/3 |
| 1228 reflections | (Δ/σ)max < 0.001 |
| 101 parameters | Δρmax = 0.55 e Å−3 |
| 10 restraints | Δρmin = −1.38 e Å−3 |
Special details
| Experimental. The crystal was coated in Exxon Paratone N hydrocarbon oil and mounted on a thin mohair fibre attached to a copper pin. Upon mounting on the diffractometer, the crystal was quenched to 150(K) under a cold nitrogen gas stream supplied by an Oxford Cryosystems Cryostream and data were collected at this temperature. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Co1 | 0.64313 (4) | 0.00064 (2) | 0.20676 (8) | 0.00402 (17) | |
| Co2 | 0.26113 (6) | 0.2500 | 0.42866 (12) | 0.00838 (19) | |
| P1 | 0.39745 (9) | 0.09462 (5) | 0.27639 (18) | 0.0123 (2) | |
| O1 | 0.5259 (3) | 0.07872 (14) | 0.1463 (7) | 0.0202 (6) | |
| O2 | 0.3026 (2) | 0.03968 (15) | 0.1432 (5) | 0.0148 (5) | |
| O3 | 0.3601 (3) | 0.17305 (16) | 0.2139 (6) | 0.0201 (7) | |
| O4 | 0.3927 (4) | 0.2500 | 0.7439 (7) | 0.0155 (8) | |
| O5 | 0.1149 (4) | 0.2500 | 0.1406 (9) | 0.0192 (8) | |
| O6 | 0.1642 (3) | 0.16927 (15) | 0.6593 (6) | 0.0197 (6) | |
| O7 | 0.4000 (4) | 0.08075 (16) | 0.5755 (6) | 0.0311 (8) | |
| H1 | 0.098 (4) | 0.161 (3) | 0.553 (10) | 0.047* | |
| H2 | 0.2194 (15) | 0.1390 (19) | 0.739 (9) | 0.047* | |
| H3 | 0.132 (8) | 0.280 (5) | 0.005 (15) | 0.047* | 0.50 |
| H4 | 0.044 (5) | 0.231 (5) | 0.204 (18) | 0.047* | 0.50 |
| H5 | 0.403 (13) | 0.296 (3) | 0.80 (3) | 0.047* | 0.50 |
| H6 | 0.366 (13) | 0.210 (4) | 0.83 (3) | 0.047* | 0.50 |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Co1 | 0.0038 (3) | 0.0055 (3) | 0.0027 (3) | 0.00152 (13) | 0.00023 (14) | 0.00025 (13) |
| Co2 | 0.0081 (3) | 0.0088 (3) | 0.0082 (3) | 0.000 | 0.0006 (2) | 0.000 |
| P1 | 0.0179 (5) | 0.0077 (4) | 0.0114 (4) | 0.0004 (3) | −0.0004 (3) | 0.0003 (3) |
| O1 | 0.0128 (13) | 0.0123 (12) | 0.0357 (16) | −0.0008 (10) | −0.0008 (12) | 0.0038 (12) |
| O2 | 0.0135 (12) | 0.0177 (12) | 0.0134 (11) | −0.0024 (10) | 0.0020 (10) | −0.0028 (10) |
| O3 | 0.0331 (17) | 0.0099 (13) | 0.0174 (14) | 0.0061 (11) | 0.0094 (11) | 0.0022 (10) |
| O4 | 0.017 (2) | 0.0169 (19) | 0.0130 (16) | 0.000 | −0.0012 (15) | 0.000 |
| O5 | 0.0136 (18) | 0.026 (2) | 0.0183 (18) | 0.000 | −0.0003 (16) | 0.000 |
| O6 | 0.0175 (14) | 0.0149 (13) | 0.0267 (14) | −0.0024 (11) | 0.0027 (12) | 0.0026 (12) |
| O7 | 0.066 (2) | 0.0131 (13) | 0.0142 (13) | −0.0105 (14) | −0.0095 (15) | 0.0020 (10) |
Geometric parameters (Å, °)
| Co1—O7i | 1.901 (3) | P1—O7 | 1.519 (3) |
| Co1—O1 | 1.918 (3) | P1—O3 | 1.521 (3) |
| Co1—O2ii | 1.983 (3) | P1—O1 | 1.537 (3) |
| Co1—O2iii | 1.986 (3) | P1—O2 | 1.570 (3) |
| Co2—O3iv | 2.058 (3) | O4—H5 | 0.893 (10) |
| Co2—O3 | 2.058 (3) | O4—H6 | 0.893 (10) |
| Co2—O4 | 2.106 (4) | O5—H3 | 0.892 (10) |
| Co2—O5 | 2.118 (4) | O5—H4 | 0.891 (10) |
| Co2—O6 | 2.138 (3) | O6—H1 | 0.89 (4) |
| Co2—O6iv | 2.138 (3) | O6—H2 | 0.90 (3) |
| O7i—Co1—O1 | 121.18 (15) | O7—P1—O3 | 111.40 (17) |
| O7i—Co1—O2ii | 105.64 (13) | O7—P1—O1 | 111.8 (2) |
| O1—Co1—O2ii | 110.16 (12) | O3—P1—O1 | 108.79 (16) |
| O7i—Co1—O2iii | 106.57 (12) | O7—P1—O2 | 108.84 (17) |
| O1—Co1—O2iii | 108.97 (13) | O3—P1—O2 | 110.43 (17) |
| O2ii—Co1—O2iii | 102.75 (8) | O1—P1—O2 | 105.46 (16) |
| O3iv—Co2—O3 | 86.26 (16) | P1—O1—Co1 | 130.42 (18) |
| O3iv—Co2—O4 | 93.09 (12) | P1—O2—Co1v | 128.01 (16) |
| O3—Co2—O4 | 93.09 (12) | P1—O2—Co1iii | 115.24 (15) |
| O3iv—Co2—O5 | 91.00 (12) | Co1v—O2—Co1iii | 116.58 (13) |
| O3—Co2—O5 | 91.00 (12) | P1—O3—Co2 | 132.08 (17) |
| O4—Co2—O5 | 174.38 (15) | Co2—O4—H5 | 108 (10) |
| O3iv—Co2—O6 | 178.01 (12) | Co2—O4—H6 | 98 (10) |
| O3—Co2—O6 | 93.16 (12) | H5—O4—H6 | 133 (3) |
| O4—Co2—O6 | 85.03 (11) | Co2—O5—H3 | 112 (6) |
| O5—Co2—O6 | 90.91 (12) | Co2—O5—H4 | 112 (6) |
| O3iv—Co2—O6iv | 93.16 (12) | H3—O5—H4 | 134 (3) |
| O3—Co2—O6iv | 178.01 (12) | Co2—O6—H1 | 100 (4) |
| O4—Co2—O6iv | 85.03 (11) | Co2—O6—H2 | 110.7 (10) |
| O5—Co2—O6iv | 90.91 (12) | H1—O6—H2 | 132 (2) |
| O6—Co2—O6iv | 87.36 (16) | P1—O7—Co1i | 133.79 (19) |
| O7—P1—O1—Co1 | 41.2 (3) | O1—P1—O2—Co1iii | −20.9 (2) |
| O3—P1—O1—Co1 | 164.7 (2) | O7—P1—O3—Co2 | −22.9 (3) |
| O2—P1—O1—Co1 | −76.9 (3) | O1—P1—O3—Co2 | −146.5 (2) |
| O7i—Co1—O1—P1 | 8.8 (3) | O2—P1—O3—Co2 | 98.2 (3) |
| O2ii—Co1—O1—P1 | −115.1 (2) | O3iv—Co2—O3—P1 | 153.57 (18) |
| O2iii—Co1—O1—P1 | 132.9 (2) | O4—Co2—O3—P1 | 60.7 (3) |
| O7—P1—O2—Co1v | 34.1 (3) | O5—Co2—O3—P1 | −115.5 (3) |
| O3—P1—O2—Co1v | −88.5 (2) | O6—Co2—O3—P1 | −24.5 (3) |
| O1—P1—O2—Co1v | 154.17 (19) | O3—P1—O7—Co1i | 143.7 (3) |
| O7—P1—O2—Co1iii | −140.96 (19) | O1—P1—O7—Co1i | −94.4 (4) |
| O3—P1—O2—Co1iii | 96.45 (18) | O2—P1—O7—Co1i | 21.7 (4) |
Symmetry codes: (i) −x+1, −y, −z+1; (ii) x+1/2, y, −z+1/2; (iii) −x+1, −y, −z; (iv) x, −y+1/2, z; (v) x−1/2, y, −z+1/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O4—H5···O3vi | 0.89 (1) | 2.25 (8) | 2.764 (4) | 116 (7) |
| O4—H5···O7iv | 0.89 (1) | 2.49 (8) | 3.209 (3) | 138 (9) |
| O4—H6···O3vii | 0.89 (1) | 2.02 (7) | 2.764 (4) | 140 (10) |
| O5—H4···O3v | 0.89 (1) | 2.25 (2) | 3.133 (5) | 170 (8) |
| O5—H4···O3viii | 0.89 (1) | 2.66 (8) | 3.133 (5) | 115 (6) |
| O6—H1···O1v | 0.90 (4) | 1.94 (3) | 2.690 (4) | 140 (4) |
| O6—H2···O7 | 0.90 (3) | 2.35 (3) | 3.008 (5) | 130 (3) |
Symmetry codes: (vi) x, −y+1/2, z+1; (iv) x, −y+1/2, z; (vii) x, y, z+1; (v) x−1/2, y, −z+1/2; (viii) x−1/2, −y+1/2, −z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: WM2193).
References
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst.37, 335–338.
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst.32, 115–119.
- Cason, C. J. (2002). POV-RAY Hallam Oaks Pty Ltd, Williamstown, Victoria, Australia.
- Cheetham, A. K., Férey, G. & Loiseau, T. (1999). Angew. Chem. Int. Ed.38, 3268–3292. [PubMed]
- Chen, Z., Gao, Q., Gao, D., Wei, Q. & Ruan, M. (2006). Mater. Lett 60, 1816–1822.
- Choudhury, A., Natarajan, S. & Rao, C. N. R. (2000). Inorg. Chem.39, 4295–4304. [DOI] [PubMed]
- Cooper, E. R., Andrews, C. D., Wheatley, P. S., Webb, P. B., Wormald, P. & Morris, R. E. (2004). Nature (London), 430, 1012–1016. [DOI] [PubMed]
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Forster, P. M., Eckert, J., Chang, J.-S., Park, J.-S., Férey, G. & Cheatham, A. K. (2003). J. Am. Chem. Soc.125, 1309–1312. [DOI] [PubMed]
- Jiang, Y. & Gao, Q. (2007). Mater. Lett.61, 2212–2216.
- Jiang, Y.-C., Lai, Y.-C., Wang, S.-L. & Lii, K.-H. (2001). Inorg. Chem.40, 5320–5321. [DOI] [PubMed]
- Kuzicki, S. M., Bell, V. A., Nair, S., Hillhouse, H. W., Jacubinas, R. M., Braunbarth, V. M., Toby, B. H. & Tsapatsis, M. (2001). Nature (London), 412, 720–724. [DOI] [PubMed]
- Lee, Y. H., Clegg, J. K., Lindoy, L. F., Lu, G. Q. M., Park, Y.-C. & Kim, Y. (2008). Acta Cryst. E64, i69–i70. [DOI] [PMC free article] [PubMed]
- Liebau, F. (1965). Acta Cryst.18, 352–354.
- Molecular Simulations (2000). WebLab ViewerPro Accelrys Software Inc., San Diego, California, USA.
- Sheldrick, G. M. (1999). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siemens (1995). SMART, SAINT and XPREP Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808028377/wm2193sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808028377/wm2193Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report