

Abstract
In the molecule of the title compound, C8H6Cl2O2, the benzene ring is oriented with respect to the planar ester group at a dihedral angle of 39.22 (3)°.
Related literature
For general background, see: Zheng et al. (2003 ▶); Al-Talib et al. (1990 ▶); Yousif et al. (1986 ▶); Ahmad et al. (2001 ▶); Al-Soud et al. (2004 ▶); El-Emam et al. (2004 ▶); Weinstock et al. (1991 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶); and of MOGUL, see: Bruno et al. (2004 ▶). 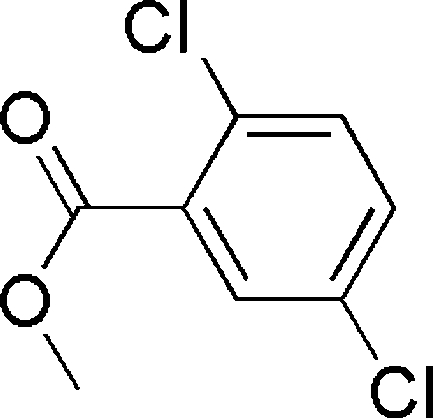
Experimental
Crystal data
C8H6Cl2O2
M r = 205.03
Triclinic,
a = 3.8452 (3) Å
b = 7.0158 (4) Å
c = 15.8510 (10) Å
α = 77.189 (6)°
β = 89.130 (7)°
γ = 83.741 (5)°
V = 414.46 (5) Å3
Z = 2
Mo Kα radiation
μ = 0.73 mm−1
T = 150 (1) K
0.68 × 0.11 × 0.06 mm
Data collection
Bruker–Nonius Kappa CCD area-detector diffractometer
Absorption correction: Gaussian (Coppens, 1970 ▶) T min = 0.864, T max = 0.971
5966 measured reflections
1840 independent reflections
1455 reflections with I > 2σ(I)
R int = 0.110
Refinement
R[F 2 > 2σ(F 2)] = 0.053
wR(F 2) = 0.146
S = 1.09
1840 reflections
109 parameters
H-atom parameters constrained
Δρmax = 0.44 e Å−3
Δρmin = −0.57 e Å−3
Data collection: COLLECT (Hooft, 1998 ▶)and DENZO (Otwinowski & Minor, 1997 ▶); cell refinement: DIRAX/LSQ (Duisenberg, 1992) ▶); data reduction: EvalCCD (Duisenberg, 1992) ▶); program(s) used to solve structure: SIR92 (Altomare et al., 1994 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2003 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks I. DOI: 10.1107/S1600536808029541/hk2533sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808029541/hk2533Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors gratefully acknowledge funds from the Higher Education Commission, Islamabad, Pakistan, and also thank the Ministry of Education of the Czech Republic for financial support (project No. VZ0021627501).
supplementary crystallographic information
Comment
The title compound is a lachrymator and a drug intermediate. Methyl 2,5-di- chlorobenzoate is widely employed in synthetic organic chemistry for example, 2,5-dichlorobenzohydrazide, 2,5-disubstituted-1,3,4-oxadiazoles (Zheng et al., 2003; Al-Talib et al., 1990) and 5-substituted-2-mercapto-1,3,4-oxadiazoles (Yousif et al., 1986; Ahmad et al., 2001; Al-Soud et al., 2004; El-Emam et al., 2004). In addition, methyl 4-(bromomethyl)benzoate has been used in the synthesis of 1-(carboxybenzyl)imidazole-5-acrylic acids, which are potent and selective angiotensin II receptor antagonists (Weinstock et al., 1991).In view of the versatility of these compounds, we have synthesized the title compound, and report herein its crystal structure.
In the molecule of the title compound, (Fig. 1), the bond lengths and angles are generally within normal ranges (Cambridge Structural Database, Version 5.28, November 2006; Mogul Version 1.1; Allen, 2002, Bruno et al., 2004). The benzene ring (C1-C6) is oriented with respect to the planar ester group (O1/O2/C1/C7/C8) at a dihedral angle of 39.22 (3)°.
Experimental
For the preparation of the title compound, the mixture of 2,5-dichlorobenzoic acid (2.05 g, 10 mmol) and absolute methanol (50 ml) in the presence of a few drops of suphuric acid was refluxed for 5 h. The excess of solvent was removed by distillation. The solid residue for filltered off, washed with water and recystallized from ethanol (30%) to give the title compound (yied; 88%, m.p. 319-321 K). Suitable single crystals of the title compound were obtained by slow evaporation of an ethanol solution at room temperature.
Refinement
H atoms were positioned geometrically, with C-H = 0.93 and 0.96 Å for aromatic and methyl H, respectively, and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(C).
Figures
Fig. 1.

The molecular structure of the title molecule, with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Reaction scheme.
Crystal data
| C8H6Cl2O2 | Z = 2 |
| Mr = 205.03 | F(000) = 208 |
| Triclinic, P1 | Dx = 1.643 Mg m−3 |
| Hall symbol: -P 1 | Melting point: 319(2) K |
| a = 3.8452 (3) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.0158 (4) Å | Cell parameters from 6024 reflections |
| c = 15.851 (1) Å | θ = 1–27.5° |
| α = 77.189 (6)° | µ = 0.73 mm−1 |
| β = 89.130 (7)° | T = 150 K |
| γ = 83.741 (5)° | Needle, colorless |
| V = 414.46 (5) Å3 | 0.68 × 0.11 × 0.06 mm |
Data collection
| Bruker–Nonius KappaCCD area-detector diffractometer | 1840 independent reflections |
| Radiation source: fine-focus sealed tube | 1455 reflections with I > 2σ(I) |
| graphite | Rint = 0.110 |
| Detector resolution: 9.091 pixels mm-1 | θmax = 27.5°, θmin = 2.6° |
| φ and ω scans | h = −4→4 |
| Absorption correction: gaussian (Coppens, 1970) | k = −9→8 |
| Tmin = 0.864, Tmax = 0.971 | l = −20→20 |
| 5966 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.053 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.146 | H-atom parameters constrained |
| S = 1.10 | w = 1/[σ2(Fo2) + (0.0527P)2 + 0.5264P] where P = (Fo2 + 2Fc2)/3 |
| 1840 reflections | (Δ/σ)max < 0.001 |
| 109 parameters | Δρmax = 0.44 e Å−3 |
| 0 restraints | Δρmin = −0.57 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.6965 (2) | 0.52269 (12) | 0.70713 (5) | 0.0309 (2) | |
| Cl2 | 1.3585 (2) | −0.28182 (13) | 0.92310 (5) | 0.0361 (3) | |
| O1 | 0.6439 (7) | −0.0150 (4) | 0.62680 (14) | 0.0347 (6) | |
| O2 | 0.8542 (6) | 0.2744 (4) | 0.57949 (13) | 0.0288 (5) | |
| C1 | 0.9066 (8) | 0.1286 (5) | 0.72871 (18) | 0.0220 (6) | |
| C2 | 0.8841 (8) | 0.2952 (5) | 0.76262 (19) | 0.0235 (6) | |
| C3 | 1.0100 (9) | 0.2857 (5) | 0.8458 (2) | 0.0278 (7) | |
| H3 | 0.9954 | 0.3983 | 0.8680 | 0.033* | |
| C4 | 1.1574 (9) | 0.1080 (5) | 0.89485 (19) | 0.0286 (7) | |
| H4 | 1.2435 | 0.1006 | 0.9501 | 0.034* | |
| C5 | 1.1755 (8) | −0.0580 (5) | 0.8613 (2) | 0.0257 (7) | |
| C6 | 1.0489 (8) | −0.0510 (5) | 0.77933 (19) | 0.0245 (6) | |
| H6 | 1.0581 | −0.1648 | 0.7581 | 0.029* | |
| C7 | 0.7825 (8) | 0.1206 (5) | 0.64006 (18) | 0.0229 (6) | |
| C8 | 0.7421 (9) | 0.2749 (5) | 0.49289 (19) | 0.0294 (7) | |
| H8A | 0.8509 | 0.1593 | 0.4761 | 0.035* | |
| H8B | 0.8089 | 0.3898 | 0.4536 | 0.035* | |
| H8C | 0.4924 | 0.2757 | 0.4914 | 0.035* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0411 (5) | 0.0249 (4) | 0.0250 (4) | 0.0029 (3) | −0.0044 (3) | −0.0047 (3) |
| Cl2 | 0.0429 (5) | 0.0328 (5) | 0.0276 (4) | 0.0027 (4) | −0.0115 (3) | 0.0014 (3) |
| O1 | 0.0496 (16) | 0.0344 (14) | 0.0231 (11) | −0.0124 (11) | −0.0066 (10) | −0.0089 (10) |
| O2 | 0.0368 (13) | 0.0354 (13) | 0.0137 (10) | −0.0082 (10) | −0.0056 (8) | −0.0017 (9) |
| C1 | 0.0214 (15) | 0.0287 (16) | 0.0156 (13) | −0.0036 (12) | −0.0005 (10) | −0.0039 (11) |
| C2 | 0.0233 (15) | 0.0256 (16) | 0.0205 (14) | −0.0043 (12) | −0.0010 (11) | −0.0021 (12) |
| C3 | 0.0331 (18) | 0.0295 (18) | 0.0230 (15) | −0.0050 (13) | −0.0007 (12) | −0.0096 (13) |
| C4 | 0.0336 (18) | 0.0348 (18) | 0.0172 (14) | −0.0055 (14) | −0.0045 (12) | −0.0042 (12) |
| C5 | 0.0233 (16) | 0.0303 (17) | 0.0206 (14) | −0.0028 (12) | −0.0014 (11) | 0.0003 (12) |
| C6 | 0.0302 (17) | 0.0236 (16) | 0.0205 (14) | −0.0008 (12) | −0.0026 (11) | −0.0073 (12) |
| C7 | 0.0257 (15) | 0.0265 (16) | 0.0166 (13) | 0.0004 (12) | −0.0013 (11) | −0.0062 (11) |
| C8 | 0.0367 (19) | 0.0368 (19) | 0.0141 (13) | 0.0003 (14) | −0.0047 (12) | −0.0061 (12) |
Geometric parameters (Å, °)
| Cl1—C2 | 1.728 (3) | C3—H3 | 0.9300 |
| Cl2—C5 | 1.737 (3) | C4—C3 | 1.382 (5) |
| O1—C7 | 1.199 (4) | C4—C5 | 1.378 (5) |
| O2—C7 | 1.326 (4) | C4—H4 | 0.9300 |
| O2—C8 | 1.444 (3) | C5—C6 | 1.384 (4) |
| C1—C2 | 1.385 (5) | C6—H6 | 0.9301 |
| C1—C6 | 1.395 (4) | C8—H8A | 0.9600 |
| C1—C7 | 1.505 (4) | C8—H8B | 0.9600 |
| C2—C3 | 1.396 (4) | C8—H8C | 0.9600 |
| C7—O2—C8 | 115.5 (3) | C4—C5—Cl2 | 119.7 (2) |
| C2—C1—C6 | 119.3 (3) | C6—C5—Cl2 | 118.9 (3) |
| C2—C1—C7 | 125.6 (3) | C5—C6—C1 | 119.3 (3) |
| C6—C1—C7 | 115.1 (3) | C5—C6—H6 | 120.4 |
| C1—C2—Cl1 | 123.1 (2) | C1—C6—H6 | 120.2 |
| C1—C2—C3 | 120.7 (3) | O1—C7—O2 | 124.7 (3) |
| C3—C2—Cl1 | 116.3 (3) | O1—C7—C1 | 122.4 (3) |
| C4—C3—C2 | 119.7 (3) | O2—C7—C1 | 112.9 (3) |
| C4—C3—H3 | 120.1 | O2—C8—H8A | 109.4 |
| C2—C3—H3 | 120.3 | O2—C8—H8B | 109.5 |
| C5—C4—C3 | 119.5 (3) | H8A—C8—H8B | 109.5 |
| C5—C4—H4 | 120.3 | O2—C8—H8C | 109.5 |
| C3—C4—H4 | 120.2 | H8A—C8—H8C | 109.5 |
| C4—C5—C6 | 121.4 (3) | H8B—C8—H8C | 109.5 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2533).
References
- Ahmad, R., Iqbal, R., Akhtar, R. H., Haq, Z. U., Duddeck, H., Stefaniak, L. & Sitkowski, J. (2001). Nucleosides Nucleotides Nucleic Acids, 20, 1671–1682. [DOI] [PubMed]
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Al-Soud, Y. A., Al-Deeri, M. N. & Al-Mosoudi, N. A. (2004). Farmaco, 59, 775–783. [DOI] [PubMed]
- Al-Talib, M., Tastoush, H. & Odeh, N. (1990). Synth. Commun.20, 1811–1814.
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst 27, 435-436.
- Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004). J. Chem. Inf. Comput. Sci.44, 2133–2144. [DOI] [PubMed]
- Coppens, P. (1970). Crystallographic Computing, edited by F. R. Ahmed, S. R. Hall & C. P. Huber, pp. 255–270. Copenhagen: Munksgaard.
- Duisenberg, A. J. M. (1992). J. Appl. Cryst.25, 92–96.
- El-Emam, A. A., Al-Deeb, O. A., Al-Omar, M. & Lehmann, J. (2004). Bioorg. Med. Chem.12, 5107–5113. [DOI] [PubMed]
- Hooft, R. (1998). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Weinstock, J. et al. (1991). J. Med. Chem.34, 1514–1517. [DOI] [PubMed]
- Yousif, M. Y., Ismail, A. M., Elman, A. A. & El-Kerdawy, M. M. (1986). J. Chem. Soc. Pak.8, 183–187.
- Zheng, X., Li, Z., Wang, Y., Chen, W., Huang, Q., Liu, C. & Song, G. (2003). J. Fluorine Chem.117, 163–169.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I. DOI: 10.1107/S1600536808029541/hk2533sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808029541/hk2533Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report