

Abstract
In the cation of the title compound, C6H7N6 +·NO3 −, the pyridine and tetrazole rings are essentially coplanar, exhibiting a dihedral angle of 6.30 (6)°. In the crystal structure, N—H⋯O, N—H⋯N, C—H⋯O and C—H⋯N hydrogen bonds form a three-dimensional network.
Related literature
For general background on the chemistry of tetrazole derivatives, see: Dunica et al. (1991 ▶); Wittenberger & Donner (1993 ▶); Zou et al. (2007 ▶); Xiong et al. (2002 ▶). For the crystal structures of related compounds, see: Dai & Fu (2008 ▶); Wang et al. (2005 ▶).
Experimental
Crystal data
C6H7N6 +·NO3 −
M r = 225.19
Monoclinic,

a = 8.3797 (17) Å
b = 6.9314 (14) Å
c = 15.881 (3) Å
β = 94.31 (3)°
V = 919.8 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.13 mm−1
T = 298 (2) K
0.30 × 0.22 × 0.20 mm
Data collection
Rigaku Mercury2 diffractometer
Absorption correction: multi-scan (CrystalClear; Rigaku, 2005 ▶) T min = 0.916, T max = 0.970
8766 measured reflections
2023 independent reflections
1520 reflections with I > 2σ(I)
R int = 0.041
Refinement
R[F 2 > 2σ(F 2)] = 0.049
wR(F 2) = 0.120
S = 1.07
2023 reflections
173 parameters
All H-atom parameters refined
Δρmax = 0.15 e Å−3
Δρmin = −0.18 e Å−3
Data collection: CrystalClear (Rigaku, 2005 ▶); cell refinement: CrystalClear; data reduction: CrystalClear; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL/PC (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL/PC.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808030791/rz2247sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808030791/rz2247Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯O2i | 0.95 (2) | 2.55 (2) | 3.328 (2) | 138.7 (18) |
| N1—H1A⋯O3i | 0.95 (2) | 1.84 (3) | 2.764 (2) | 163 (2) |
| N5—H5⋯O1ii | 0.91 (2) | 2.11 (2) | 2.989 (2) | 163 (2) |
| N5—H5⋯O3ii | 0.91 (2) | 2.21 (2) | 2.908 (2) | 133.3 (19) |
| N6—H6A⋯O1iii | 0.88 (3) | 2.31 (3) | 3.074 (3) | 145 (2) |
| N6—H6B⋯O2iii | 0.90 (3) | 2.48 (3) | 2.908 (2) | 110 (2) |
| N6—H6B⋯N2iv | 0.90 (3) | 2.32 (3) | 3.176 (3) | 158 (3) |
| C1—H1⋯O3ii | 0.96 (2) | 2.60 (2) | 3.124 (2) | 114.4 (16) |
| C1—H1⋯O2i | 0.96 (2) | 2.38 (2) | 3.308 (2) | 161.5 (18) |
| C3—H3⋯N4v | 0.95 (2) | 2.55 (2) | 3.305 (3) | 136.3 (16) |
Symmetry codes: (i)  ; (ii)
; (ii) 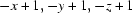 ; (iii)
; (iii) 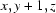 ; (iv)
; (iv)  ; (v)
; (v) 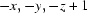 .
.
Acknowledgments
This work was supported by a Start-up Grant from Southeast University to Professor R.-G. Xiong.
supplementary crystallographic information
Comment
The tetrazole functional group has found a wide range of applications in coordination chemistry as ligand, in medicinal chemistry as a metabolically stable surrogate for the carboxylic acid group, and in materials science as high density energy material (Wang et al., 2005; Xiong et al., 2002; Zou et al., 2007; Dunica et al., 1991; Wittenberger & Donner, 1993). We report here the crystal structure of the title compound, 5-(1H-tetrazol-5-yl)pyridin-2-amine-1-ium nitrate.
In the cation of the title compound (Fig. 1) the pyridine and tetrazole rings are essentially coplanar with a dihedral angle of only 6.30 (6)°. Bond distances and angles of the tetrazole ring are within the usual range (Wang et al., 2005; Dai & Fu, 2008). The pyridine N atom is protonated. The crystal packing is consolidated by N—H···O, N—H···N, C—H···O and C—H···N hydrogen bonds to form a three-dimentional network. (Table 1, Fig. 2).
Experimental
2-Amino-5-cyanopyridine (30 mmol), NaN3 (45 mmol), NH4Cl (33 mmol) and DMF (50 ml) were added in a flask under nitrogen atmosphere and the mixture stirred at 110°C for 20 h. The resulting solution was then poured into ice-water (100 ml), and a white solid was obtained after adding nitrate acid (6 M) till pH=6. The precipitate was filtered and washed with distilled water. Colourless block-shaped crystals suitable for X-ray analysis were obtained from the crude product by slow evaporation of an ethanol/nitric acid (50:1 v/v) solution.
Refinement
All H atoms were located in difference Fourier maps and refined freely.
Figures
Fig. 1.

A view of the title compound with the atomic numbering scheme. Displacement ellipsoids were drawn at the 30% probability level.
Fig. 2.

The crystal packing of the title compound viewed along the b axis showing the three-dimensionnal hydrogen bonding network (dashed lines). Hydrogen atoms not involved in hydrogen bonding are omitted for clarity.
Crystal data
| C6H7N6+·NO3− | F(000) = 464 |
| Mr = 225.19 | Dx = 1.626 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1772 reflections |
| a = 8.3797 (17) Å | θ = 2.4–27.1° |
| b = 6.9314 (14) Å | µ = 0.13 mm−1 |
| c = 15.881 (3) Å | T = 298 K |
| β = 94.31 (3)° | Block, colourless |
| V = 919.8 (3) Å3 | 0.30 × 0.22 × 0.20 mm |
| Z = 4 |
Data collection
| Rigaku Mercury2 (2x2 bin mode) diffractometer | 2023 independent reflections |
| Radiation source: fine-focus sealed tube | 1520 reflections with I > 2σ(I) |
| graphite | Rint = 0.041 |
| Detector resolution: 13.6612 pixels mm-1 | θmax = 27.1°, θmin = 3.2° |
| ω scans | h = −10→10 |
| Absorption correction: multi-scan (CrystalClear; Rigaku, 2005) | k = −8→8 |
| Tmin = 0.916, Tmax = 0.970 | l = −20→20 |
| 8766 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.049 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.120 | All H-atom parameters refined |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0527P)2 + 0.2064P] where P = (Fo2 + 2Fc2)/3 |
| 2023 reflections | (Δ/σ)max < 0.001 |
| 173 parameters | Δρmax = 0.15 e Å−3 |
| 0 restraints | Δρmin = −0.18 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.49052 (18) | 0.0891 (2) | 0.58329 (9) | 0.0552 (4) | |
| O2 | 0.3288 (2) | 0.0420 (2) | 0.68089 (9) | 0.0632 (5) | |
| O3 | 0.46287 (18) | 0.3054 (2) | 0.67890 (10) | 0.0588 (4) | |
| N7 | 0.42643 (19) | 0.1424 (2) | 0.64808 (10) | 0.0406 (4) | |
| N1 | 0.2311 (2) | 0.0535 (2) | 0.27752 (10) | 0.0416 (4) | |
| C2 | 0.2037 (2) | 0.2485 (3) | 0.40803 (11) | 0.0359 (4) | |
| C5 | 0.2593 (2) | 0.5628 (3) | 0.51712 (11) | 0.0401 (4) | |
| N5 | 0.3153 (2) | 0.5556 (2) | 0.43967 (10) | 0.0427 (4) | |
| C4 | 0.1695 (2) | 0.4035 (3) | 0.54193 (12) | 0.0411 (5) | |
| C6 | 0.1714 (2) | 0.0831 (3) | 0.35207 (11) | 0.0367 (4) | |
| N4 | 0.0781 (2) | −0.0636 (3) | 0.36762 (11) | 0.0526 (5) | |
| N6 | 0.2911 (3) | 0.7171 (3) | 0.56527 (13) | 0.0550 (5) | |
| C3 | 0.1443 (2) | 0.2500 (3) | 0.48954 (12) | 0.0399 (4) | |
| N2 | 0.1724 (2) | −0.1144 (2) | 0.24520 (10) | 0.0497 (5) | |
| C1 | 0.2875 (2) | 0.4049 (3) | 0.38512 (12) | 0.0414 (5) | |
| N3 | 0.0808 (2) | −0.1835 (3) | 0.29971 (11) | 0.0577 (5) | |
| H6A | 0.345 (3) | 0.815 (4) | 0.5462 (15) | 0.066 (8)* | |
| H6B | 0.260 (4) | 0.723 (4) | 0.618 (2) | 0.092 (10)* | |
| H1A | 0.306 (3) | 0.126 (3) | 0.2484 (15) | 0.066 (7)* | |
| H4 | 0.132 (2) | 0.408 (3) | 0.5949 (13) | 0.045 (5)* | |
| H3 | 0.085 (3) | 0.143 (3) | 0.5074 (13) | 0.052 (6)* | |
| H1 | 0.325 (3) | 0.423 (3) | 0.3298 (14) | 0.056 (6)* | |
| H5 | 0.370 (3) | 0.658 (3) | 0.4211 (14) | 0.061 (7)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0669 (10) | 0.0599 (10) | 0.0416 (8) | 0.0075 (8) | 0.0238 (7) | −0.0058 (7) |
| O2 | 0.0879 (12) | 0.0556 (9) | 0.0497 (9) | −0.0239 (9) | 0.0292 (9) | −0.0047 (7) |
| O3 | 0.0629 (10) | 0.0531 (9) | 0.0633 (10) | −0.0168 (8) | 0.0236 (8) | −0.0186 (7) |
| N7 | 0.0451 (9) | 0.0430 (9) | 0.0345 (8) | 0.0031 (7) | 0.0084 (7) | −0.0002 (7) |
| N1 | 0.0519 (10) | 0.0426 (9) | 0.0314 (8) | −0.0025 (8) | 0.0094 (7) | 0.0003 (7) |
| C2 | 0.0373 (10) | 0.0387 (10) | 0.0321 (9) | −0.0009 (8) | 0.0055 (7) | 0.0016 (8) |
| C5 | 0.0445 (10) | 0.0413 (11) | 0.0344 (9) | 0.0030 (8) | 0.0015 (8) | −0.0005 (8) |
| N5 | 0.0499 (10) | 0.0397 (9) | 0.0391 (9) | −0.0064 (8) | 0.0081 (8) | 0.0028 (7) |
| C4 | 0.0457 (11) | 0.0481 (11) | 0.0302 (9) | −0.0006 (9) | 0.0077 (8) | 0.0010 (8) |
| C6 | 0.0395 (10) | 0.0406 (10) | 0.0306 (9) | −0.0012 (8) | 0.0063 (8) | 0.0029 (7) |
| N4 | 0.0623 (11) | 0.0533 (11) | 0.0440 (10) | −0.0183 (9) | 0.0154 (9) | −0.0049 (8) |
| N6 | 0.0739 (14) | 0.0439 (11) | 0.0475 (11) | −0.0070 (10) | 0.0060 (10) | −0.0057 (9) |
| C3 | 0.0414 (10) | 0.0445 (11) | 0.0344 (10) | −0.0052 (9) | 0.0076 (8) | 0.0037 (8) |
| N2 | 0.0650 (11) | 0.0454 (10) | 0.0392 (9) | −0.0052 (9) | 0.0077 (8) | −0.0030 (8) |
| C1 | 0.0469 (11) | 0.0453 (11) | 0.0331 (10) | −0.0040 (9) | 0.0101 (8) | 0.0021 (8) |
| N3 | 0.0750 (13) | 0.0515 (11) | 0.0475 (10) | −0.0159 (10) | 0.0118 (9) | −0.0056 (8) |
Geometric parameters (Å, °)
| O1—N7 | 1.2517 (19) | N5—C1 | 1.366 (2) |
| O2—N7 | 1.221 (2) | N5—H5 | 0.91 (2) |
| O3—N7 | 1.260 (2) | C4—C3 | 1.358 (3) |
| N1—C6 | 1.336 (2) | C4—H4 | 0.92 (2) |
| N1—N2 | 1.350 (2) | C6—N4 | 1.317 (2) |
| N1—H1A | 0.95 (2) | N4—N3 | 1.363 (2) |
| C2—C1 | 1.356 (3) | N6—H6A | 0.88 (3) |
| C2—C3 | 1.422 (2) | N6—H6B | 0.90 (3) |
| C2—C6 | 1.463 (3) | C3—H3 | 0.95 (2) |
| C5—N6 | 1.330 (3) | N2—N3 | 1.292 (2) |
| C5—N5 | 1.350 (2) | C1—H1 | 0.96 (2) |
| C5—C4 | 1.409 (3) | ||
| O2—N7—O1 | 121.71 (17) | C5—C4—H4 | 117.3 (12) |
| O2—N7—O3 | 119.76 (16) | N4—C6—N1 | 108.35 (17) |
| O1—N7—O3 | 118.53 (16) | N4—C6—C2 | 125.21 (16) |
| C6—N1—N2 | 108.63 (16) | N1—C6—C2 | 126.44 (17) |
| C6—N1—H1A | 131.0 (14) | C6—N4—N3 | 106.09 (16) |
| N2—N1—H1A | 120.3 (14) | C5—N6—H6A | 120.5 (16) |
| C1—C2—C3 | 117.54 (18) | C5—N6—H6B | 120.9 (19) |
| C1—C2—C6 | 122.64 (16) | H6A—N6—H6B | 119 (2) |
| C3—C2—C6 | 119.82 (17) | C4—C3—C2 | 120.98 (18) |
| N6—C5—N5 | 119.03 (19) | C4—C3—H3 | 119.4 (13) |
| N6—C5—C4 | 123.88 (19) | C2—C3—H3 | 119.6 (13) |
| N5—C5—C4 | 117.09 (17) | N3—N2—N1 | 106.44 (16) |
| C5—N5—C1 | 123.48 (17) | C2—C1—N5 | 120.56 (17) |
| C5—N5—H5 | 119.1 (15) | C2—C1—H1 | 123.9 (13) |
| C1—N5—H5 | 117.4 (15) | N5—C1—H1 | 115.4 (13) |
| C3—C4—C5 | 120.32 (18) | N2—N3—N4 | 110.49 (17) |
| C3—C4—H4 | 122.4 (13) | ||
| N6—C5—N5—C1 | −179.14 (19) | C2—C6—N4—N3 | 179.73 (19) |
| C4—C5—N5—C1 | 0.9 (3) | C5—C4—C3—C2 | −1.7 (3) |
| N6—C5—C4—C3 | −179.0 (2) | C1—C2—C3—C4 | 0.6 (3) |
| N5—C5—C4—C3 | 1.0 (3) | C6—C2—C3—C4 | −178.50 (18) |
| N2—N1—C6—N4 | 0.7 (2) | C6—N1—N2—N3 | −0.4 (2) |
| N2—N1—C6—C2 | −179.74 (18) | C3—C2—C1—N5 | 1.2 (3) |
| C1—C2—C6—N4 | −173.07 (19) | C6—C2—C1—N5 | −179.71 (18) |
| C3—C2—C6—N4 | 6.0 (3) | C5—N5—C1—C2 | −2.0 (3) |
| C1—C2—C6—N1 | 7.5 (3) | N1—N2—N3—N4 | 0.0 (2) |
| C3—C2—C6—N1 | −173.47 (18) | C6—N4—N3—N2 | 0.5 (2) |
| N1—C6—N4—N3 | −0.7 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O2i | 0.95 (2) | 2.55 (2) | 3.328 (2) | 138.7 (18) |
| N1—H1A···O3i | 0.95 (2) | 1.84 (3) | 2.764 (2) | 163 (2) |
| N5—H5···O1ii | 0.91 (2) | 2.11 (2) | 2.989 (2) | 163 (2) |
| N5—H5···O3ii | 0.91 (2) | 2.21 (2) | 2.908 (2) | 133.3 (19) |
| N6—H6A···O1iii | 0.88 (3) | 2.31 (3) | 3.074 (3) | 145 (2) |
| N6—H6B···O2iii | 0.90 (3) | 2.48 (3) | 2.908 (2) | 110 (2) |
| N6—H6B···N2iv | 0.90 (3) | 2.32 (3) | 3.176 (3) | 158 (3) |
| C1—H1···O3ii | 0.96 (2) | 2.60 (2) | 3.124 (2) | 114.4 (16) |
| C1—H1···O2i | 0.96 (2) | 2.38 (2) | 3.308 (2) | 161.5 (18) |
| C3—H3···N4v | 0.95 (2) | 2.55 (2) | 3.305 (3) | 136.3 (16) |
Symmetry codes: (i) x, −y+1/2, z−1/2; (ii) −x+1, −y+1, −z+1; (iii) x, y+1, z; (iv) x, −y+1/2, z+1/2; (v) −x, −y, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: RZ2247).
References
- Dai, W. & Fu, D.-W. (2008). Acta Cryst. E64, o1444. [DOI] [PMC free article] [PubMed]
- Dunica, J. V., Pierce, M. E. & Santella, J. B. III (1991). J. Org. Chem.56, 2395–2400.
- Rigaku (2005). CrystalClear Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, X.-S., Tang, Y.-Z., Huang, X.-F., Qu, Z.-R., Che, C.-M., Chan, C. W. H. & Xiong, R.-G. (2005). Inorg. Chem.44, 5278–5285. [DOI] [PubMed]
- Wittenberger, S. J. & Donner, B. G. (1993). J. Org. Chem.58, 4139–4141.
- Xiong, R.-G., Xue, X., Zhao, H., You, X.-Z., Abrahams, B. F. & Xue, Z.-L. (2002). Angew. Chem. Int. Ed.41, 3800–3803. [DOI] [PubMed]
- Zou, Y., Hong, S., Park, M., Chun, H. & Lah, M. S. (2007). Chem. Commun.28, 5182–5184. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808030791/rz2247sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808030791/rz2247Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report