

Abstract
In the structure of the title compound, [PdCl2(C5H5N)2], the PdII atom is located on an inversion centre and the pyridine rings are coplanar. There is intermolecular π–π stacking between the pyridyl rings, with a centroid-to-centroid separation of 3.916 (1) Å. The structure is a new polymorph of two previously determined structures [Viossat, Dung & Robert (1993 ▶). Acta Cryst. C49, 84–85; Liao & Lee (2006 ▶). Acta Cryst. E62, m680–m681].
Related literature
For the other two polymorphs of the title compound, see: Viossat et al. (1993 ▶); Liao & Lee (2006 ▶).
Experimental
Crystal data
[PdCl2(C5H5N)2]
M r = 335.50
Monoclinic,
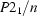
a = 3.9159 (2) Å
b = 8.7921 (4) Å
c = 16.2974 (8) Å
β = 90.442 (3)°
V = 561.09 (5) Å3
Z = 2
Mo Kα radiation
μ = 2.10 mm−1
T = 150 (2) K
0.45 × 0.10 × 0.07 mm
Data collection
Bruker SMART APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2003 ▶) T min = 0.452, T max = 0.867
5861 measured reflections
1445 independent reflections
1314 reflections with I > 2σ
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.023
wR(F 2) = 0.056
S = 1.10
1445 reflections
70 parameters
H-atom parameters constrained
Δρmax = 0.91 e Å−3
Δρmin = −1.03 e Å−3
Data collection: APEX2 (Bruker, 2004 ▶); cell refinement: SAINT (Bruker, 2004 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808033552/bi2308sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033552/bi2308Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors are grateful to the National Science Council of Taiwan for financial support.
supplementary crystallographic information
Comment
Two polymorphic forms of the title compound have already been determined (Viossat et al., 1993; Liao & Lee, 2006). The polymorphic form determined by us previously has a plate-like crystal habit (Liao & Lee, 2006). Herein, we present a new polymorphic form of the title compound. This new form has a rod-like habit. The PdII atom, situated at a centre of inversion, has a square-planer coordination geometry with two trans pyridine ligands and two trans chloride ligands (Fig. 1). Similar to the polymorph determined by us previously (Liao & Lee, 2006), in this new polymorphic form the two pyridine rings are co-planar. The co-planariity in these two forms is in sharp contrast to that in the other polymorph in which the the two pyridine planes make an angle of 160.0 (5)° (Viossat et al., 1993).
The crystal packing is distinctly different in the three polymorphs. A view of the packing arrangement for the new polymorphic form is shown in Fig. 2. Intermolecular π–π stacking exists between the pyridyl rings, with centroid–centroid separation 3.916 Å.
Experimental
The title compound is commercially available. Crystals were grown by slow diffusion of diethyl ether into a dimethylformamide solution containing the compound. The polymorphic form has a rod-like crystal habit.
Refinement
All H atoms could be identified in the difference Fourier map, but were positioned geometrically and refined as riding atoms, with C—H = 0.95 Å and Uiso(H) = 1.2Ueq(C).
Figures
Fig. 1.

Molecular structure of the title compound, showing 50% displacement ellipsoids for non-H atoms. The H atoms are dipicted by circles of an arbitrary radius. The unlabelled atoms are related to the labelled ones by -x, 1 - y, -z.
Fig. 2.

Packing diagram of the title compound viewed along the a axis.
Crystal data
| [PdCl2(C5H5N)2] | F(000) = 328 |
| Mr = 335.50 | Dx = 1.986 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 4043 reflections |
| a = 3.9159 (2) Å | θ = 2.5–34.2° |
| b = 8.7921 (4) Å | µ = 2.10 mm−1 |
| c = 16.2974 (8) Å | T = 150 K |
| β = 90.442 (3)° | Rod, colourless |
| V = 561.09 (5) Å3 | 0.45 × 0.10 × 0.07 mm |
| Z = 2 |
Data collection
| Bruker SMART APEXII diffractometer | 1445 independent reflections |
| Radiation source: fine-focus sealed tube | 1314 reflections with I > 2σ |
| graphite | Rint = 0.028 |
| ω scans | θmax = 28.7°, θmin = 2.6° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2003) | h = −5→5 |
| Tmin = 0.452, Tmax = 0.867 | k = −11→8 |
| 5861 measured reflections | l = −22→19 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.023 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.056 | H-atom parameters constrained |
| S = 1.10 | w = 1/[σ2(Fo2) + (0.0105P)2 + 1.3011P] where P = (Fo2 + 2Fc2)/3 |
| 1445 reflections | (Δ/σ)max < 0.001 |
| 70 parameters | Δρmax = 0.91 e Å−3 |
| 0 restraints | Δρmin = −1.03 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.1177 (6) | 0.6747 (3) | 0.15154 (15) | 0.0203 (5) | |
| H1 | 0.2253 | 0.5833 | 0.1690 | 0.024* | |
| C2 | 0.1019 (7) | 0.7956 (3) | 0.20522 (16) | 0.0249 (5) | |
| H2 | 0.1976 | 0.7878 | 0.2588 | 0.030* | |
| C3 | −0.0560 (7) | 0.9288 (3) | 0.17976 (17) | 0.0251 (5) | |
| H3 | −0.0737 | 1.0130 | 0.2160 | 0.030* | |
| C4 | −0.1875 (7) | 0.9376 (3) | 0.10081 (18) | 0.0238 (5) | |
| H4 | −0.2913 | 1.0288 | 0.0817 | 0.029* | |
| C5 | −0.1655 (6) | 0.8117 (3) | 0.05015 (15) | 0.0194 (5) | |
| H5 | −0.2595 | 0.8169 | −0.0037 | 0.023* | |
| Cl1 | 0.24377 (16) | 0.64576 (7) | −0.10193 (4) | 0.01983 (13) | |
| N1 | −0.0149 (5) | 0.6822 (2) | 0.07511 (12) | 0.0159 (4) | |
| Pd1 | 0.0000 | 0.5000 | 0.0000 | 0.01359 (8) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0236 (11) | 0.0198 (12) | 0.0174 (11) | 0.0000 (9) | −0.0019 (9) | −0.0002 (9) |
| C2 | 0.0263 (12) | 0.0314 (14) | 0.0171 (12) | −0.0021 (11) | −0.0009 (10) | −0.0051 (10) |
| C3 | 0.0272 (13) | 0.0227 (13) | 0.0256 (13) | −0.0032 (11) | 0.0024 (10) | −0.0096 (10) |
| C4 | 0.0252 (12) | 0.0163 (12) | 0.0298 (14) | 0.0018 (10) | 0.0006 (10) | −0.0020 (10) |
| C5 | 0.0232 (11) | 0.0170 (11) | 0.0180 (11) | −0.0001 (9) | −0.0011 (9) | 0.0003 (9) |
| Cl1 | 0.0255 (3) | 0.0174 (3) | 0.0167 (3) | −0.0023 (2) | 0.0026 (2) | 0.0009 (2) |
| N1 | 0.0199 (9) | 0.0140 (9) | 0.0138 (9) | −0.0011 (7) | 0.0000 (7) | −0.0012 (7) |
| Pd1 | 0.01822 (13) | 0.01113 (12) | 0.01142 (12) | 0.00025 (9) | −0.00070 (8) | −0.00068 (8) |
Geometric parameters (Å, °)
| C1—N1 | 1.347 (3) | C4—H4 | 0.950 |
| C1—C2 | 1.379 (4) | C5—N1 | 1.344 (3) |
| C1—H1 | 0.950 | C5—H5 | 0.950 |
| C2—C3 | 1.386 (4) | Cl1—Pd1 | 2.3104 (6) |
| C2—H2 | 0.950 | N1—Pd1 | 2.017 (2) |
| C3—C4 | 1.385 (4) | Pd1—N1i | 2.017 (2) |
| C3—H3 | 0.950 | Pd1—Cl1i | 2.3104 (6) |
| C4—C5 | 1.384 (4) | ||
| N1—C1—C2 | 122.0 (2) | N1—C5—C4 | 121.8 (2) |
| N1—C1—H1 | 119.0 | N1—C5—H5 | 119.1 |
| C2—C1—H1 | 119.0 | C4—C5—H5 | 119.1 |
| C1—C2—C3 | 118.9 (2) | C5—N1—C1 | 119.1 (2) |
| C1—C2—H2 | 120.5 | C5—N1—Pd1 | 120.25 (16) |
| C3—C2—H2 | 120.5 | C1—N1—Pd1 | 120.64 (17) |
| C4—C3—C2 | 119.2 (2) | N1i—Pd1—N1 | 180.0 |
| C4—C3—H3 | 120.4 | N1i—Pd1—Cl1 | 89.43 (6) |
| C2—C3—H3 | 120.4 | N1—Pd1—Cl1 | 90.57 (6) |
| C5—C4—C3 | 119.0 (2) | N1i—Pd1—Cl1i | 90.57 (6) |
| C5—C4—H4 | 120.5 | N1—Pd1—Cl1i | 89.42 (6) |
| C3—C4—H4 | 120.5 | Cl1—Pd1—Cl1i | 180.0 |
Symmetry codes: (i) −x, −y+1, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BI2308).
References
- Bruker (2004). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Liao, C.-Y. & Lee, H. M. (2006). Acta Cryst. E62, m680–m681.
- Sheldrick, G. M. (2003). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Viossat, B., Dung, N.-H. & Robert, F. (1993). Acta Cryst. C49, 84–85.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808033552/bi2308sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033552/bi2308Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report