

Abstract
The approximately planar molecule of the title compound, C9H11N3O2S, is linked to adjacent molecules by O—H⋯S hydrogen bonds to form a zigzag chain. Adjacent chains are consolidated by N—H⋯O hydrogen bonds into a two-dimensional array. An intramolecular O—H⋯N link is also present.
Related literature
For the structure of isomeric 2,5-dihydroxybenzaldehyde 4-methylthiosemicarbazone, see: Tan et al. (2008 ▶).
Experimental
Crystal data
C9H11N3O2S
M r = 225.27
Monoclinic,

a = 18.0046 (6) Å
b = 4.6436 (1) Å
c = 12.2842 (4) Å
β = 106.695 (2)°
V = 983.74 (5) Å3
Z = 4
Mo Kα radiation
μ = 0.31 mm−1
T = 100 (2) K
0.09 × 0.06 × 0.03 mm
Data collection
Bruker SMART APEX diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.973, T max = 0.991
4390 measured reflections
2128 independent reflections
1925 reflections with I > 2σ(I)
R int = 0.034
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.109
S = 1.11
2128 reflections
153 parameters
6 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.31 e Å−3
Δρmin = −0.22 e Å−3
Absolute structure: Flack (1983 ▶), with 814 Friedel pairs
Flack parameter: 0.00 (1)
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: publCIF (Westrip, 2008 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808033308/tk2316sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033308/tk2316Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1O⋯N1 | 0.84 (1) | 1.93 (3) | 2.694 (3) | 151 (6) |
| O2—H2O⋯S1i | 0.84 (1) | 2.54 (1) | 3.365 (2) | 170 (4) |
| N2—H2N⋯O1ii | 0.87 (1) | 2.11 (1) | 2.950 (4) | 162 (3) |
Symmetry codes: (i) 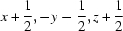 ; (ii)
; (ii)  .
.
Acknowledgments
We thank the University of Malaya (PJP FS316/2008 C) for supporting this study. KWT thanks the Ministry of Higher Education for an SLAI scholarship.
supplementary crystallographic information
Comment
In continuation of on-going studies into the structural chemistry of thiosemicarbazones (Tan et al., 2008), the title compound (I) was investigated. Molecule (I), Fig. 1, is essentially planar and is consolidated into a 2-D array by a combination of N-H···O and O-H···S hydrogen bonding contacts, Table 1.
Experimental
4-Methylthiosemicarbazide (0.11 g, 1 mmol) and 2,4-dihydroxybenzaldehyde (0.14 g, 1 mmol) were heated in ethanol (10 ml) for 1 h. Slow evaporation of the solvent yielded yellow crystals.
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.95 to 0.98 Å) and were included in the refinement in the riding model approximation, with Uiso>(H) set to 1.2-1.5 Ueq(C). The hydroxy and amino H-atoms were located in a difference Fourier map, and were refined with distance retraints of O–H = 0.84±0.01 and N–H = 0.88±0.01 Å; their temperature factors were freely refined.
Figures
Fig. 1.

Thermal ellipsoid (Barbour, 2001) plot of (I) drawn at the 70% probability level. Hydrogen atoms are drawn as spheres of arbitrary radii.
Crystal data
| C9H11N3O2S | F(000) = 472 |
| Mr = 225.27 | Dx = 1.521 Mg m−3 |
| Monoclinic, Cc | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: C -2yc | Cell parameters from 1090 reflections |
| a = 18.0046 (6) Å | θ = 2.4–24.9° |
| b = 4.6436 (1) Å | µ = 0.31 mm−1 |
| c = 12.2842 (4) Å | T = 100 K |
| β = 106.695 (2)° | Prims, yellow |
| V = 983.74 (5) Å3 | 0.09 × 0.06 × 0.03 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX diffractometer | 2128 independent reflections |
| Radiation source: fine-focus sealed tube | 1925 reflections with I > 2σ(I) |
| graphite | Rint = 0.034 |
| ω scans | θmax = 27.5°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −23→22 |
| Tmin = 0.973, Tmax = 0.991 | k = −6→6 |
| 4390 measured reflections | l = −15→15 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.038 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.109 | w = 1/[σ2(Fo2) + (0.0598P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.11 | (Δ/σ)max = 0.001 |
| 2128 reflections | Δρmax = 0.31 e Å−3 |
| 153 parameters | Δρmin = −0.22 e Å−3 |
| 6 restraints | Absolute structure: Flack (1983), 814 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: 0.00 (1) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.50003 (5) | 0.63599 (16) | 0.50001 (6) | 0.01776 (19) | |
| O1 | 0.65168 (12) | −0.1181 (5) | 0.93136 (19) | 0.0174 (5) | |
| O2 | 0.84529 (13) | −0.8242 (5) | 1.05294 (19) | 0.0198 (5) | |
| N1 | 0.63927 (15) | 0.1424 (5) | 0.7309 (2) | 0.0144 (5) | |
| N2 | 0.60440 (15) | 0.2962 (6) | 0.6322 (2) | 0.0147 (5) | |
| N3 | 0.53164 (15) | 0.5567 (6) | 0.7241 (2) | 0.0166 (6) | |
| C1 | 0.73692 (17) | −0.2114 (7) | 0.8141 (2) | 0.0128 (6) | |
| C2 | 0.71210 (17) | −0.2660 (7) | 0.9113 (2) | 0.0122 (6) | |
| C3 | 0.74812 (18) | −0.4725 (7) | 0.9891 (3) | 0.0141 (6) | |
| H3 | 0.7300 | −0.5117 | 1.0530 | 0.017* | |
| C4 | 0.81112 (16) | −0.6232 (6) | 0.9739 (2) | 0.0142 (6) | |
| C5 | 0.83798 (18) | −0.5674 (7) | 0.8800 (3) | 0.0174 (7) | |
| H5 | 0.8815 | −0.6688 | 0.8704 | 0.021* | |
| C6 | 0.80096 (17) | −0.3645 (7) | 0.8017 (3) | 0.0159 (7) | |
| H6 | 0.8192 | −0.3274 | 0.7378 | 0.019* | |
| C7 | 0.69685 (17) | −0.0131 (7) | 0.7250 (2) | 0.0142 (6) | |
| H7 | 0.7142 | 0.0020 | 0.6590 | 0.017* | |
| C8 | 0.54711 (17) | 0.4887 (7) | 0.6279 (3) | 0.0149 (6) | |
| C9 | 0.47552 (19) | 0.7743 (8) | 0.7317 (3) | 0.0206 (7) | |
| H9A | 0.4879 | 0.8460 | 0.8099 | 0.031* | |
| H9B | 0.4234 | 0.6899 | 0.7100 | 0.031* | |
| H9C | 0.4773 | 0.9340 | 0.6804 | 0.031* | |
| H1O | 0.633 (3) | −0.017 (11) | 0.874 (3) | 0.08 (2)* | |
| H2O | 0.8796 (19) | −0.909 (8) | 1.032 (4) | 0.038 (13)* | |
| H2N | 0.6097 (19) | 0.218 (7) | 0.5703 (17) | 0.011 (8)* | |
| H3N | 0.5552 (19) | 0.476 (7) | 0.7898 (17) | 0.018 (9)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0196 (4) | 0.0180 (4) | 0.0138 (3) | 0.0027 (4) | 0.0018 (3) | 0.0021 (4) |
| O1 | 0.0193 (13) | 0.0174 (12) | 0.0159 (11) | 0.0038 (9) | 0.0059 (9) | 0.0029 (10) |
| O2 | 0.0216 (12) | 0.0196 (13) | 0.0171 (12) | 0.0069 (10) | 0.0037 (9) | 0.0040 (9) |
| N1 | 0.0159 (13) | 0.0126 (13) | 0.0125 (12) | −0.0018 (11) | 0.0008 (10) | 0.0013 (10) |
| N2 | 0.0176 (13) | 0.0163 (13) | 0.0097 (13) | 0.0024 (11) | 0.0030 (10) | −0.0001 (11) |
| N3 | 0.0195 (14) | 0.0144 (13) | 0.0152 (13) | 0.0023 (11) | 0.0037 (10) | 0.0014 (10) |
| C1 | 0.0153 (14) | 0.0135 (15) | 0.0103 (14) | −0.0029 (12) | 0.0048 (11) | −0.0005 (12) |
| C2 | 0.0105 (14) | 0.0147 (15) | 0.0129 (15) | −0.0017 (12) | 0.0056 (11) | −0.0034 (12) |
| C3 | 0.0162 (15) | 0.0159 (16) | 0.0120 (15) | −0.0032 (12) | 0.0069 (12) | −0.0012 (11) |
| C4 | 0.0148 (16) | 0.0139 (14) | 0.0113 (14) | −0.0007 (12) | −0.0006 (12) | 0.0000 (12) |
| C5 | 0.0155 (16) | 0.0163 (16) | 0.0180 (17) | 0.0012 (12) | 0.0010 (12) | −0.0031 (12) |
| C6 | 0.0121 (15) | 0.0214 (18) | 0.0134 (15) | −0.0031 (14) | 0.0023 (12) | −0.0033 (13) |
| C7 | 0.0157 (16) | 0.0164 (16) | 0.0117 (15) | −0.0035 (13) | 0.0058 (12) | −0.0016 (12) |
| C8 | 0.0154 (15) | 0.0127 (15) | 0.0175 (16) | −0.0031 (12) | 0.0064 (12) | 0.0002 (12) |
| C9 | 0.0169 (16) | 0.0266 (19) | 0.0201 (17) | 0.0003 (13) | 0.0079 (13) | −0.0038 (13) |
Geometric parameters (Å, °)
| S1—C8 | 1.699 (3) | C1—C2 | 1.413 (4) |
| O1—C2 | 1.367 (4) | C1—C7 | 1.454 (4) |
| O1—H1O | 0.838 (10) | C2—C3 | 1.378 (4) |
| O2—C4 | 1.360 (4) | C3—C4 | 1.391 (4) |
| O2—H2O | 0.836 (10) | C3—H3 | 0.9500 |
| N1—C7 | 1.283 (4) | C4—C5 | 1.397 (4) |
| N1—N2 | 1.392 (3) | C5—C6 | 1.374 (4) |
| N2—C8 | 1.354 (4) | C5—H5 | 0.9500 |
| N2—H2N | 0.871 (10) | C6—H6 | 0.9500 |
| N3—C8 | 1.328 (4) | C7—H7 | 0.9500 |
| N3—C9 | 1.451 (4) | C9—H9A | 0.9800 |
| N3—H3N | 0.880 (10) | C9—H9B | 0.9800 |
| C1—C6 | 1.400 (4) | C9—H9C | 0.9800 |
| C2—O1—H1O | 106 (4) | C3—C4—C5 | 120.5 (3) |
| C4—O2—H2O | 109 (3) | C6—C5—C4 | 119.5 (3) |
| C7—N1—N2 | 114.1 (3) | C6—C5—H5 | 120.3 |
| C8—N2—N1 | 121.4 (3) | C4—C5—H5 | 120.3 |
| C8—N2—H2N | 121 (2) | C5—C6—C1 | 121.4 (3) |
| N1—N2—H2N | 114 (2) | C5—C6—H6 | 119.3 |
| C8—N3—C9 | 123.4 (3) | C1—C6—H6 | 119.3 |
| C8—N3—H3N | 123 (3) | N1—C7—C1 | 123.2 (3) |
| C9—N3—H3N | 114 (3) | N1—C7—H7 | 118.4 |
| C6—C1—C2 | 118.1 (3) | C1—C7—H7 | 118.4 |
| C6—C1—C7 | 119.1 (3) | N3—C8—N2 | 118.3 (3) |
| C2—C1—C7 | 122.8 (3) | N3—C8—S1 | 123.4 (2) |
| O1—C2—C3 | 117.7 (3) | N2—C8—S1 | 118.3 (2) |
| O1—C2—C1 | 121.6 (3) | N3—C9—H9A | 109.5 |
| C3—C2—C1 | 120.8 (3) | N3—C9—H9B | 109.5 |
| C2—C3—C4 | 119.8 (3) | H9A—C9—H9B | 109.5 |
| C2—C3—H3 | 120.1 | N3—C9—H9C | 109.5 |
| C4—C3—H3 | 120.1 | H9A—C9—H9C | 109.5 |
| O2—C4—C3 | 117.9 (3) | H9B—C9—H9C | 109.5 |
| O2—C4—C5 | 121.6 (3) | ||
| C7—N1—N2—C8 | −174.4 (3) | C4—C5—C6—C1 | 0.2 (5) |
| C6—C1—C2—O1 | 177.7 (3) | C2—C1—C6—C5 | 1.4 (4) |
| C7—C1—C2—O1 | −4.9 (4) | C7—C1—C6—C5 | −176.0 (3) |
| C6—C1—C2—C3 | −2.5 (4) | N2—N1—C7—C1 | −174.0 (3) |
| C7—C1—C2—C3 | 174.8 (3) | C6—C1—C7—N1 | −177.7 (3) |
| O1—C2—C3—C4 | −178.3 (3) | C2—C1—C7—N1 | 5.0 (5) |
| C1—C2—C3—C4 | 1.9 (5) | C9—N3—C8—N2 | 175.6 (3) |
| C2—C3—C4—O2 | 179.7 (3) | C9—N3—C8—S1 | −3.1 (4) |
| C2—C3—C4—C5 | −0.2 (5) | N1—N2—C8—N3 | 8.8 (4) |
| O2—C4—C5—C6 | 179.3 (3) | N1—N2—C8—S1 | −172.5 (2) |
| C3—C4—C5—C6 | −0.9 (5) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O···N1 | 0.84 (1) | 1.93 (3) | 2.694 (3) | 151 (6) |
| O2—H2O···S1i | 0.84 (1) | 2.54 (1) | 3.365 (2) | 170 (4) |
| N2—H2N···O1ii | 0.87 (1) | 2.11 (1) | 2.950 (4) | 162 (3) |
Symmetry codes: (i) x+1/2, −y−1/2, z+1/2; (ii) x, −y, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: TK2316).
References
- Barbour, L. J. (2001). J. Supramol. Chem.1, 189–191.
- Bruker (2007). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tan, K. W., Ng, C. H., Maah, M. J. & Ng, S. W. (2008). Acta Cryst. E64, o1344. [DOI] [PMC free article] [PubMed]
- Westrip, S. P. (2008). publCIF In preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808033308/tk2316sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033308/tk2316Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report