

Abstract
The asymmetric unit of the title compound, C17H17N2O2 +·Cl−·H2O, contains one-half of the cation, one-half of a water molecule and a chloride anion. The complete cation is generated by crystallographic two-fold symmetry, with one C atom lying on the rotation axis. The O and Cl atoms have site symmetry 2. The imidazolidium ring is oriented at a dihedral angle of 4.15 (3)° with respect to the 4-methoxyphenyl ring and an intramolecular C—H⋯O interaction occurs. In the crystal structure, intermolecular O—H⋯Cl and C—H⋯Cl hydrogen bonds link the molecules. There is a π–π contact between the imidazolidium and 4-methoxyphenyl rings [centroid-to-centroid distance = 3.625(3 Å]. There is also a C—H⋯π contact between the methyl group and the 4-methoxyphenyl ring.
Related literature
For general background, see: Lin & Vasam (2005 ▶). For bond-length data, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C17H17N2O2 +·Cl−·H2O
M r = 334.79
Monoclinic,

a = 15.6706 (19) Å
b = 9.4198 (9) Å
c = 5.4026 (4) Å
β = 90.156 (1)°
V = 797.50 (14) Å3
Z = 2
Mo Kα radiation
μ = 0.26 mm−1
T = 298 (2) K
0.20 × 0.11 × 0.09 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.951, T max = 0.977
2026 measured reflections
749 independent reflections
688 reflections with I > 2σ(I)
R int = 0.021
Refinement
R[F 2 > 2σ(F 2)] = 0.029
wR(F 2) = 0.076
S = 1.01
749 reflections
107 parameters
1 restraint
H-atom parameters constrained
Δρmax = 0.11 e Å−3
Δρmin = −0.22 e Å−3
Data collection: SMART (Bruker, 1998 ▶); cell refinement: SAINT (Bruker, 1999 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶) and PLATON (Spek, 2003 ▶); software used to prepare material for publication: SHELXTL (Sheldrick, 2008 ▶) and PLATON.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808033965/hk2542sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033965/hk2542Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg2 is the centroid of the C3–C8 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H2⋯Cl1 | 0.85 | 2.29 | 3.133 (3) | 173 |
| C1—H1⋯O2 | 0.93 | 2.13 | 3.060 (3) | 180 |
| C2—H2A⋯Cl1i | 0.93 | 2.69 | 3.474 (3) | 142 |
| C4—H4⋯O2 | 0.93 | 2.47 | 3.391 (3) | 170 |
| C9—H9C⋯Cg2ii | 0.96 | 2.91 | 3.629 (3) | 133 |
Symmetry codes: (i)  ; (ii)
; (ii) 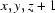 .
.
Acknowledgments
The authors are grateful to the National Natural Science Foundation of China (grant No. 20772103), the Natural Science Foundation of Jiangsu Province (grant No. BK 2007028) and the Surpassing Project of Jiangsu Province (grant No. CX07S_016z) for financial support.
supplementary crystallographic information
Comment
Imidazole and its derivatives such as imidazolium cation are important compounds playing important roles in medical, organic and material chemistry (Lin & Vasam, 2005). A broad application of imidazolium now is to synthesize ionic liquids. Recently, ionic liquids are attracting much attention as alternative reaction media for synthesis and catalysis. Its applications in many different areas including separation processes, catalyst, electrochemistry, electrolytes in solar cells and lubricants are widely recognized. Therefore, the need of ionic liquids with specific chemical and physical properties become stronger. We report herein the synthesis and crystal structure of the title compound.
The asymmetric unit of the title compound (Fig. 1) contains one half-molecule, one half-water molecule and a chloride atom. The bond lengths (Allen et al., 1987) and angles are within normal ranges. Rings A (N1/N1'/C1/C2/C2') and B (C3–C8) are, of course, planar and the dihedral angle between them is A/B = 4.15 (3)° [symmetry code: (') -x, y, -z]. Intramolecular C—H···O and O—H···Cl hydrogen bonds (Table 1) link the molecules.
In the crystal structure, intramolecular C—H···O and O—H···Cl and intermolecular C—H···Cl hydrogen bonds (Table 1) link the molecules (Fig. 2), in which they may be effective in the stabilization of the structure. The π–π contact between the imidazolidium and 4-methoxyphenyl rings, Cg1···Cg2i [symmetry code: (i) 1 - x, y, 1 - z, where Cg1 and Cg2 are the centroids of the rings A (N1/N1'/C1/C2/C2') and B (C3–C8), respectively] may further stabilize the structure, with centroid-centroid distance of 3.625 (3) Å. There also exist a C—H···π contact (Table 1) between the methyl group and the 4-methoxyphenyl ring.
Experimental
The reaction of 4-methoxybenzenamine (2 mmol) with formaldehyde (aq. 37%, 1 mmol) and glyoxal (aq. 40%, 1 mmol) in ethanol (95%) at 273–278 K for 8 h afforded 1-(2,3-diethoxy-4-(4-methoxyphenyl)cyclopentyl)-4-methoxybenzene (yield; 89%). The title compound was obtained through the oxidization of 1-(2,3-diethoxy-4-(4-methoxyphenyl)cyclopentyl)-4-methoxybenzene by phosgene in DMF at 268–273 K (yield 95%).
Refinement
H atoms were positioned geometrically, with O—H = 0.85 Å (for H2O) and C—H = 0.93 and 0.96 Å for aromatic and methyl H, respectively, and constrained to ride on their parent atoms with Uiso(H) = xUeq(C,O), where x = 1.5 for methyl H and x = 1.2 for all other H atoms.
Figures
Fig. 1.

The molecular structure of the title molecule, with the atom-numbering scheme [symmetry code: (') -x, y, -z].
Fig. 2.

A partial packing diagram. Hydrogen bonds are shown as dashed lines.
Crystal data
| C17H17N2O2+·Cl−·H2O | F(000) = 352 |
| Mr = 334.79 | Dx = 1.394 Mg m−3 |
| Monoclinic, C2 | Melting point = 492–494 K |
| Hall symbol: -C 2y | Mo Kα radiation, λ = 0.71073 Å |
| a = 15.6706 (19) Å | Cell parameters from 1340 reflections |
| b = 9.4198 (9) Å | θ = 2.5–28.3° |
| c = 5.4026 (4) Å | µ = 0.26 mm−1 |
| β = 90.156 (1)° | T = 298 K |
| V = 797.50 (14) Å3 | Block, colourless |
| Z = 2 | 0.20 × 0.11 × 0.09 mm |
Data collection
| Bruker SMART CCD area-detector diffractometer | 749 independent reflections |
| Radiation source: fine-focus sealed tube | 688 reflections with I > 2σ(I) |
| graphite | Rint = 0.021 |
| φ and ω scans | θmax = 25.0°, θmin = 2.5° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −18→13 |
| Tmin = 0.951, Tmax = 0.977 | k = −9→11 |
| 2026 measured reflections | l = −6→6 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.029 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.076 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.0503P)2 + 0.1521P] where P = (Fo2 + 2Fc2)/3 |
| 749 reflections | (Δ/σ)max < 0.001 |
| 107 parameters | Δρmax = 0.11 e Å−3 |
| 1 restraint | Δρmin = −0.22 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.5000 | 0.84927 (12) | 0.5000 | 0.0800 (5) | |
| O1 | 0.24434 (13) | 0.4740 (2) | 0.9080 (3) | 0.0519 (5) | |
| O2 | 0.5000 | 0.6808 (3) | 0.0000 | 0.0567 (8) | |
| H2 | 0.5000 | 0.7335 | 0.1277 | 0.068* | |
| N1 | 0.45802 (12) | 0.27396 (19) | 0.1594 (3) | 0.0334 (5) | |
| C1 | 0.5000 | 0.3560 (4) | 0.0000 | 0.0350 (7) | |
| H1 | 0.5000 | 0.4547 | 0.0000 | 0.042* | |
| C2 | 0.47435 (17) | 0.1342 (3) | 0.0983 (5) | 0.0444 (6) | |
| H2A | 0.4534 | 0.0545 | 0.1795 | 0.053* | |
| C3 | 0.40340 (14) | 0.3225 (3) | 0.3567 (4) | 0.0335 (5) | |
| C4 | 0.39574 (17) | 0.4670 (3) | 0.4036 (5) | 0.0420 (6) | |
| H4 | 0.4262 | 0.5322 | 0.3094 | 0.050* | |
| C5 | 0.34270 (17) | 0.5135 (3) | 0.5903 (5) | 0.0449 (6) | |
| H5 | 0.3378 | 0.6101 | 0.6225 | 0.054* | |
| C6 | 0.29642 (15) | 0.4164 (3) | 0.7309 (5) | 0.0386 (6) | |
| C7 | 0.30558 (17) | 0.2728 (3) | 0.6859 (5) | 0.0446 (6) | |
| H7 | 0.2758 | 0.2073 | 0.7812 | 0.054* | |
| C8 | 0.35934 (16) | 0.2262 (3) | 0.4979 (5) | 0.0440 (6) | |
| H8 | 0.3654 | 0.1296 | 0.4678 | 0.053* | |
| C9 | 0.19340 (19) | 0.3780 (4) | 1.0497 (5) | 0.0580 (8) | |
| H9A | 0.1588 | 0.3219 | 0.9402 | 0.087* | |
| H9B | 0.1573 | 0.4309 | 1.1594 | 0.087* | |
| H9C | 0.2299 | 0.3168 | 1.1447 | 0.087* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.1591 (13) | 0.0423 (5) | 0.0387 (5) | 0.000 | 0.0069 (6) | 0.000 |
| O1 | 0.0545 (11) | 0.0522 (12) | 0.0491 (11) | 0.0011 (9) | 0.0158 (9) | 0.0059 (9) |
| O2 | 0.092 (2) | 0.0344 (15) | 0.0434 (14) | 0.000 | 0.0109 (14) | 0.000 |
| N1 | 0.0365 (11) | 0.0278 (10) | 0.0358 (10) | −0.0015 (8) | −0.0027 (8) | 0.0024 (8) |
| C1 | 0.0400 (18) | 0.0271 (15) | 0.0379 (15) | 0.000 | 0.0009 (14) | 0.000 |
| C2 | 0.0572 (16) | 0.0300 (13) | 0.0461 (13) | −0.0031 (11) | 0.0030 (11) | 0.0012 (11) |
| C3 | 0.0319 (12) | 0.0362 (13) | 0.0323 (10) | −0.0010 (10) | −0.0018 (9) | 0.0024 (10) |
| C4 | 0.0473 (15) | 0.0333 (14) | 0.0455 (14) | −0.0024 (11) | 0.0086 (11) | 0.0080 (11) |
| C5 | 0.0510 (15) | 0.0339 (13) | 0.0499 (14) | 0.0029 (12) | 0.0085 (11) | 0.0012 (12) |
| C6 | 0.0356 (14) | 0.0442 (15) | 0.0360 (12) | −0.0008 (11) | 0.0001 (11) | 0.0027 (11) |
| C7 | 0.0480 (15) | 0.0421 (16) | 0.0438 (14) | −0.0081 (12) | 0.0045 (11) | 0.0072 (12) |
| C8 | 0.0518 (16) | 0.0319 (13) | 0.0484 (15) | −0.0044 (12) | 0.0012 (13) | 0.0019 (12) |
| C9 | 0.0485 (16) | 0.069 (2) | 0.0566 (16) | −0.0036 (14) | 0.0137 (13) | 0.0109 (15) |
Geometric parameters (Å, °)
| O1—C6 | 1.371 (3) | C4—C5 | 1.380 (4) |
| O1—C9 | 1.430 (3) | C4—H4 | 0.9300 |
| O2—H2 | 0.8500 | C5—C6 | 1.394 (4) |
| N1—C1 | 1.332 (3) | C5—H5 | 0.9300 |
| N1—C2 | 1.382 (3) | C6—C7 | 1.382 (4) |
| N1—C3 | 1.443 (3) | C7—C8 | 1.392 (4) |
| C1—N1i | 1.332 (3) | C7—H7 | 0.9300 |
| C1—H1 | 0.9300 | C8—H8 | 0.9300 |
| C2—C2i | 1.334 (5) | C9—H9A | 0.9600 |
| C2—H2A | 0.9300 | C9—H9B | 0.9600 |
| C3—C8 | 1.372 (3) | C9—H9C | 0.9600 |
| C3—C4 | 1.390 (4) | ||
| C6—O1—C9 | 117.2 (2) | C4—C5—H5 | 119.8 |
| C1—N1—C2 | 107.8 (2) | C6—C5—H5 | 119.8 |
| C1—N1—C3 | 126.1 (2) | O1—C6—C7 | 124.9 (2) |
| C2—N1—C3 | 126.1 (2) | O1—C6—C5 | 115.6 (2) |
| N1i—C1—N1 | 109.1 (3) | C7—C6—C5 | 119.5 (2) |
| N1i—C1—H1 | 125.5 | C6—C7—C8 | 120.0 (2) |
| N1—C1—H1 | 125.5 | C6—C7—H7 | 120.0 |
| C2i—C2—N1 | 107.63 (14) | C8—C7—H7 | 120.0 |
| C2i—C2—H2A | 126.2 | C3—C8—C7 | 120.2 (2) |
| N1—C2—H2A | 126.2 | C3—C8—H8 | 119.9 |
| C8—C3—C4 | 120.1 (2) | C7—C8—H8 | 119.9 |
| C8—C3—N1 | 120.1 (2) | O1—C9—H9A | 109.5 |
| C4—C3—N1 | 119.8 (2) | O1—C9—H9B | 109.5 |
| C5—C4—C3 | 119.8 (2) | H9A—C9—H9B | 109.5 |
| C5—C4—H4 | 120.1 | O1—C9—H9C | 109.5 |
| C3—C4—H4 | 120.1 | H9A—C9—H9C | 109.5 |
| C4—C5—C6 | 120.4 (2) | H9B—C9—H9C | 109.5 |
| C2—N1—C1—N1i | 0.12 (13) | C3—C4—C5—C6 | −0.4 (4) |
| C3—N1—C1—N1i | −178.4 (2) | C9—O1—C6—C7 | −2.5 (4) |
| C1—N1—C2—C2i | −0.3 (3) | C9—O1—C6—C5 | 177.7 (2) |
| C3—N1—C2—C2i | 178.2 (2) | C4—C5—C6—O1 | −178.7 (2) |
| C1—N1—C3—C8 | 175.65 (18) | C4—C5—C6—C7 | 1.5 (4) |
| C2—N1—C3—C8 | −2.6 (3) | O1—C6—C7—C8 | 178.9 (2) |
| C1—N1—C3—C4 | −4.5 (3) | C5—C6—C7—C8 | −1.3 (4) |
| C2—N1—C3—C4 | 177.3 (2) | C4—C3—C8—C7 | 1.1 (3) |
| C8—C3—C4—C5 | −0.9 (4) | N1—C3—C8—C7 | −179.0 (2) |
| N1—C3—C4—C5 | 179.2 (2) | C6—C7—C8—C3 | 0.0 (4) |
Symmetry codes: (i) −x+1, y, −z.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2···Cl1 | 0.85 | 2.29 | 3.133 (3) | 173 |
| C1—H1···O2 | 0.93 | 2.13 | 3.060 (3) | 180 |
| C2—H2A···Cl1ii | 0.93 | 2.69 | 3.474 (3) | 142 |
| C4—H4···O2 | 0.93 | 2.47 | 3.391 (3) | 170 |
| C9—H9C···Cg2iii | 0.96 | 2.91 | 3.629 (3) | 133 |
Symmetry codes: (ii) x+1/2, y−1/2, z; (iii) x, y, z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2542).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bruker (1998). SMART Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (1999). SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Lin, I. J. B. & Vasam, C. S. (2005). J. Organomet. Chem.690, 3498–3512.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808033965/hk2542sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808033965/hk2542Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report