

Abstract
In the molecule of the title compound, C11H15NO4S, the S atom environment is distorted tetrahedral. The methoxycarbonyl group is oriented at a dihedral angle of 11.8 (2)° with respect to the benzene ring. In the crystal structure, intermolecular C—H⋯O hydrogen bonds link the molecules into centrosymmetric dimers.
Related literature
For general background, see: Reissenweber & Mangold (1982 ▶); Mookherjee et al. (1989 ▶); Tadashi et al. (1982 ▶). For related literature, see: Siddiqui et al. (2006 ▶,2007a
▶,b
▶); Lombardino (1972 ▶); Hanson & Hitchcook (2004 ▶).
Experimental
Crystal data
C11H15NO4S
M r = 257.30
Triclinic,
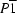
a = 8.0161 (4) Å
b = 8.4386 (4) Å
c = 10.5329 (5) Å
α = 85.244 (3)°
β = 78.721 (3)°
γ = 62.650 (3)°
V = 620.61 (5) Å3
Z = 2
Mo Kα radiation
μ = 0.26 mm−1
T = 296 (2) K
0.25 × 0.18 × 0.12 mm
Data collection
Bruker Kappa APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005) T min = 0.935, T max = 0.958
11895 measured reflections
3012 independent reflections
1817 reflections with I > 3σ(I)
R int = 0.032
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.123
S = 1.00
3012 reflections
154 parameters
H-atom parameters constrained
Δρmax = 0.37 e Å−3
Δρmin = −0.36 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: APEX2; data reduction: SAINT (Bruker, 2007 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶) and PLATON (Spek, 2003 ▶); software used to prepare material for publication: WinGX publication routines (Farrugia, 1999 ▶) and PLATON.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680800007X/hk2412sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680800007X/hk2412Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected geometric parameters (Å, °).
| S1—O1 | 1.421 (2) |
| S1—O2 | 1.4303 (19) |
| S1—N1 | 1.6269 (18) |
| S1—C9 | 1.747 (2) |
| O1—S1—O2 | 119.88 (12) |
| O1—S1—N1 | 107.29 (11) |
| O2—S1—N1 | 107.34 (11) |
| O1—S1—C9 | 108.32 (13) |
| O2—S1—C9 | 106.53 (13) |
| N1—S1—C9 | 106.83 (11) |
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C9—H9B⋯O1i | 0.96 | 2.50 | 3.372 (5) | 151 |
| C7—H7B⋯O3 | 0.97 | 2.57 | 3.044 (4) | 110 |
| C9—H9C⋯O3 | 0.96 | 2.59 | 3.152 (4) | 117 |
Symmetry code: (i) 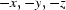 .
.
Acknowledgments
The authors acknowledge the Higher Education Commision, Islamabad, Pakistan, and Bana International, Karachi, Pakistan, for funding the purchase of the diffractometer and for technical support, respectively.
supplementary crystallographic information
Comment
Alkyl anthranilates are valuable starting materials for the preparation of pesticides, dyes and drugs (Reissenweber & Mangold, 1982). They find organoleptic uses in augmenting the aroma or taste of perfume compositions, colognes, perfumed articles, foodstuffs, medicinal products and in enhancing the effects of deodorancy (Mookherjee et al., 1989). Particularly, the N-substituted thioesters of anthranilic acid have been found potent inhibitors of the serine proteases human leukocyte (HL) elastase, porcine pancreatic elastase, cathepsin G, and bovine chymotrypsin Aα (Tadashi et al., 1982). In continuation of our research program to synthesize new biologically important 1,2-benzothiazine 1,1-dioxide molecules (Siddiqui et al., 2006; Siddiqui et al., 2007a,b), we embarked on the syntheses of 2,1-benzothiazine 2,2-dioxide, as well. Contrary to the N-methyl substituent (Lombardino, 1972), the N-ethyl derivative is being reported for the first time.
The title compound, (I), is an important precursor for the synthesis of 2,1-benzothiazine 2,2-dioxide molecule. The structure determination of (I) is undertaken in order to understand the conformational geometry around the sulfur and nitrogen atoms, due to the addition of ethyl group.
In the molecule of (I), (Fig. 1) S1 atom adopts a distorted tetrahedral coordination geometry with two O, one N and one C atoms of methylsulfonyl amino group (Table 1). The sulfur bonds are shortened, while the bond angles aroud it are different with respect to the corresponding values reported in methyl anthranilate N-methanesulfonamide, (II), (Hanson & Hitchcook, 2004).
In (I), the addition of ethyl group at N1, instead of H-atom in (II), has changed the orientation of CH3 group and O4 atom as compared with (II). On the other hand, the O3—C10 [1.193 (3) Å] and O4—C10 [1.328 (3) Å] bonds in (I), are reported as 1.2202 (16) Å and 1.3324 (16) Å, respectively, in (II). The methyl carboxylate group (O3/O4/C10/C11) is oriented at a dihedral angle of 11.8 (2)° with respect to the phenyl ring (C1—C6).
In the crystal structure, intermolecular C—H···O hydrogen bonds (Table 2) link the molecules into centrosymmetric dimers (Fig. 2), in which they may be effective in the stabilization of the structure.
Experimental
For the preparation of the title compound, the suspension of hexane-washed sodium hydride (50% in mineral oil) was prepared in dry dimethylformamide (3 ml). A solution of methyl N-methylsulfonylanthranilate (70 mg, 0.306 mmol) in dry dimethylformamide (5 ml) was added to the suspension (76 mg, 0.368 mmol), and stirred for 45 min at room temperature. Then, a solution of ethyl iodide (144 mg, 0.92 mmol) in ether (5 ml) was added to it. The resulting white suspension was stirred for 1.5 h and poured into hydrochloric acid (3 N, 50 ml) to produce a yellow suspension which was extracted with chloroform (4 × 25 ml). The combined extract was dried over calcium sulfate and evaporated under reduced pressure (11 torr) to get the title compound (yield; 58 mg, 73%, m.p. 330–331 K). Crystals suitable for X-ray analysis were obtained by slow evaporation of CHCl3.
Refinement
H atoms were positioned geometrically, with C—H = 0.93, 0.97 and 0.96 Å for aromatic, methylene and methyl H, and constrained to ride on their parent atoms, with Uiso(H) = xUeq(C), where x = 1.5 for methyl H, and x = 1.2 for all other H atoms.
Figures
Fig. 1.

The molecular structure of the title molecule, with the atom-numbering scheme. Displacement ellipsoids are drawn at the 30% probability level.
Fig. 2.

A partial packing diagram of (I). Hydrogen bonds are shown as dashed lines [symmetry code: (a) -x, -y, -z].
Crystal data
| C11H15NO4S | Z = 2 |
| Mr = 257.30 | F000 = 272 |
| Triclinic, P1 | Dx = 1.377 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation λ = 0.71073 Å |
| a = 8.0161 (4) Å | Cell parameters from 3596 reflections |
| b = 8.4386 (4) Å | θ = 2.7–24.8º |
| c = 10.5329 (5) Å | µ = 0.26 mm−1 |
| α = 85.244 (3)º | T = 296 (2) K |
| β = 78.721 (3)º | Prismatic, colourless |
| γ = 62.650 (3)º | 0.25 × 0.18 × 0.12 mm |
| V = 620.61 (5) Å3 |
Data collection
| Bruker KAPPA APEXII CCD diffractometer | 3012 independent reflections |
| Radiation source: fine-focus sealed tube | 1817 reflections with I > 3σ(I) |
| Monochromator: graphite | Rint = 0.032 |
| Detector resolution: 8.33 pixels mm-1 | θmax = 28.3º |
| T = 296(2) K | θmin = 2.0º |
| ω scans | h = −10→10 |
| Absorption correction: multi-scan(SADABS; Bruker, 2005) | k = −11→11 |
| Tmin = 0.935, Tmax = 0.958 | l = −14→14 |
| 11895 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.043 | H-atom parameters constrained |
| wR(F2) = 0.123 | w = 1/[σ2(Fo2) + (0.0727P)2 + 0.1532P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.00 | (Δ/σ)max < 0.001 |
| 3012 reflections | Δρmax = 0.37 e Å−3 |
| 154 parameters | Δρmin = −0.36 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.14595 (10) | 0.13878 (8) | 0.07989 (5) | 0.0507 (2) | |
| O1 | −0.0510 (3) | 0.1954 (3) | 0.08167 (19) | 0.0688 (6) | |
| O2 | 0.2655 (3) | 0.1495 (3) | −0.03718 (16) | 0.0707 (6) | |
| O3 | 0.2630 (3) | 0.0251 (3) | 0.40332 (17) | 0.0811 (7) | |
| O4 | 0.0983 (3) | 0.1413 (3) | 0.59441 (16) | 0.0700 (6) | |
| N1 | 0.1604 (3) | 0.2545 (3) | 0.18962 (17) | 0.0457 (5) | |
| C1 | −0.0041 (3) | 0.3538 (3) | 0.2846 (2) | 0.0443 (6) | |
| C2 | −0.0200 (3) | 0.3048 (3) | 0.4158 (2) | 0.0436 (6) | |
| C3 | −0.1825 (4) | 0.4142 (4) | 0.5006 (3) | 0.0605 (7) | |
| H3 | −0.195 | 0.3845 | 0.5878 | 0.073* | |
| C4 | −0.3251 (4) | 0.5646 (4) | 0.4595 (4) | 0.0758 (9) | |
| H4 | −0.4337 | 0.6344 | 0.5181 | 0.091* | |
| C5 | −0.3076 (5) | 0.6121 (4) | 0.3320 (4) | 0.0801 (10) | |
| H5 | −0.4039 | 0.7146 | 0.3039 | 0.096* | |
| C6 | −0.1476 (4) | 0.5080 (4) | 0.2458 (3) | 0.0654 (8) | |
| H6 | −0.1356 | 0.542 | 0.1596 | 0.078* | |
| C7 | 0.3401 (4) | 0.2613 (4) | 0.1897 (2) | 0.0565 (7) | |
| H7A | 0.4445 | 0.1594 | 0.1417 | 0.068* | |
| H7B | 0.3614 | 0.2524 | 0.2781 | 0.068* | |
| C8 | 0.3412 (5) | 0.4294 (5) | 0.1310 (3) | 0.0872 (11) | |
| H8A | 0.4615 | 0.427 | 0.1332 | 0.131* | |
| H8B | 0.2402 | 0.5307 | 0.1794 | 0.131* | |
| H8C | 0.3228 | 0.4378 | 0.0429 | 0.131* | |
| C9 | 0.2485 (4) | −0.0838 (3) | 0.1283 (2) | 0.0573 (7) | |
| H9A | 0.3819 | −0.1245 | 0.1276 | 0.086* | |
| H9B | 0.2333 | −0.1556 | 0.0697 | 0.086* | |
| H9C | 0.1866 | −0.0938 | 0.2141 | 0.086* | |
| C10 | 0.1286 (4) | 0.1421 (3) | 0.4659 (2) | 0.0457 (6) | |
| C11 | 0.2403 (5) | −0.0050 (4) | 0.6546 (3) | 0.0754 (9) | |
| H11A | 0.204 | 0.0075 | 0.747 | 0.113* | |
| H11B | 0.3615 | −0.0042 | 0.6282 | 0.113* | |
| H11C | 0.2499 | −0.1157 | 0.6285 | 0.113* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0718 (5) | 0.0469 (4) | 0.0342 (3) | −0.0235 (3) | −0.0201 (3) | 0.0011 (2) |
| O1 | 0.0720 (13) | 0.0634 (12) | 0.0718 (12) | −0.0208 (10) | −0.0347 (10) | −0.0126 (9) |
| O2 | 0.1094 (17) | 0.0694 (13) | 0.0324 (9) | −0.0411 (12) | −0.0106 (9) | 0.0037 (8) |
| O3 | 0.0977 (16) | 0.0587 (12) | 0.0408 (10) | 0.0047 (11) | −0.0141 (10) | −0.0030 (9) |
| O4 | 0.0661 (13) | 0.0920 (15) | 0.0355 (9) | −0.0220 (11) | −0.0101 (9) | 0.0021 (9) |
| N1 | 0.0579 (13) | 0.0471 (11) | 0.0337 (9) | −0.0239 (10) | −0.0109 (9) | −0.0018 (8) |
| C1 | 0.0527 (15) | 0.0365 (12) | 0.0475 (12) | −0.0200 (11) | −0.0169 (11) | −0.0023 (9) |
| C2 | 0.0484 (14) | 0.0446 (13) | 0.0417 (12) | −0.0224 (12) | −0.0104 (11) | −0.0062 (10) |
| C3 | 0.0547 (17) | 0.0681 (18) | 0.0569 (15) | −0.0256 (15) | −0.0035 (13) | −0.0186 (13) |
| C4 | 0.0540 (19) | 0.065 (2) | 0.095 (2) | −0.0127 (16) | −0.0083 (17) | −0.0345 (17) |
| C5 | 0.067 (2) | 0.0508 (17) | 0.106 (3) | −0.0015 (15) | −0.038 (2) | −0.0169 (17) |
| C6 | 0.079 (2) | 0.0450 (15) | 0.0677 (17) | −0.0174 (15) | −0.0307 (16) | 0.0023 (12) |
| C7 | 0.0632 (17) | 0.0686 (17) | 0.0439 (13) | −0.0347 (15) | −0.0087 (12) | −0.0046 (12) |
| C8 | 0.122 (3) | 0.094 (3) | 0.075 (2) | −0.077 (2) | −0.013 (2) | 0.0089 (18) |
| C9 | 0.0779 (19) | 0.0455 (14) | 0.0483 (14) | −0.0252 (14) | −0.0173 (13) | −0.0010 (11) |
| C10 | 0.0556 (16) | 0.0491 (14) | 0.0348 (11) | −0.0255 (13) | −0.0077 (11) | −0.0016 (10) |
| C11 | 0.082 (2) | 0.095 (2) | 0.0440 (14) | −0.0339 (18) | −0.0227 (15) | 0.0166 (14) |
Geometric parameters (Å, °)
| S1—O1 | 1.421 (2) | C4—H4 | 0.93 |
| S1—O2 | 1.4303 (19) | C5—C6 | 1.372 (4) |
| S1—N1 | 1.6269 (18) | C5—H5 | 0.93 |
| S1—C9 | 1.747 (2) | C6—H6 | 0.93 |
| O3—C10 | 1.193 (3) | C7—C8 | 1.503 (4) |
| O4—C10 | 1.328 (3) | C7—H7A | 0.97 |
| O4—C11 | 1.443 (3) | C7—H7B | 0.97 |
| N1—C1 | 1.431 (3) | C8—H8A | 0.96 |
| N1—C7 | 1.468 (3) | C8—H8B | 0.96 |
| C1—C6 | 1.380 (3) | C8—H8C | 0.96 |
| C1—C2 | 1.408 (3) | C9—H9A | 0.96 |
| C2—C3 | 1.387 (3) | C9—H9B | 0.96 |
| C2—C10 | 1.485 (3) | C9—H9C | 0.96 |
| C3—C4 | 1.369 (4) | C11—H11A | 0.96 |
| C3—H3 | 0.93 | C11—H11B | 0.96 |
| C4—C5 | 1.368 (5) | C11—H11C | 0.96 |
| O1—S1—O2 | 119.88 (12) | N1—C7—C8 | 112.8 (2) |
| O1—S1—N1 | 107.29 (11) | N1—C7—H7A | 109 |
| O2—S1—N1 | 107.34 (11) | C8—C7—H7A | 109 |
| O1—S1—C9 | 108.32 (13) | N1—C7—H7B | 109 |
| O2—S1—C9 | 106.53 (13) | C8—C7—H7B | 109 |
| N1—S1—C9 | 106.83 (11) | H7A—C7—H7B | 107.8 |
| C10—O4—C11 | 116.5 (2) | C7—C8—H8A | 109.5 |
| C1—N1—C7 | 119.82 (18) | C7—C8—H8B | 109.5 |
| C1—N1—S1 | 119.87 (16) | H8A—C8—H8B | 109.5 |
| C7—N1—S1 | 120.29 (16) | C7—C8—H8C | 109.5 |
| C6—C1—C2 | 119.3 (2) | H8A—C8—H8C | 109.5 |
| C6—C1—N1 | 118.0 (2) | H8B—C8—H8C | 109.5 |
| C2—C1—N1 | 122.6 (2) | S1—C9—H9A | 109.5 |
| C3—C2—C1 | 117.9 (2) | S1—C9—H9B | 109.5 |
| C3—C2—C10 | 119.3 (2) | H9A—C9—H9B | 109.5 |
| C1—C2—C10 | 122.8 (2) | S1—C9—H9C | 109.5 |
| C4—C3—C2 | 121.8 (3) | H9A—C9—H9C | 109.5 |
| C4—C3—H3 | 119.1 | H9B—C9—H9C | 109.5 |
| C2—C3—H3 | 119.1 | O3—C10—O4 | 121.8 (2) |
| C5—C4—C3 | 119.9 (3) | O3—C10—C2 | 126.8 (2) |
| C5—C4—H4 | 120 | O4—C10—C2 | 111.4 (2) |
| C3—C4—H4 | 120 | O4—C11—H11A | 109.5 |
| C4—C5—C6 | 119.7 (3) | O4—C11—H11B | 109.5 |
| C4—C5—H5 | 120.1 | H11A—C11—H11B | 109.5 |
| C6—C5—H5 | 120.1 | O4—C11—H11C | 109.5 |
| C5—C6—C1 | 121.3 (3) | H11A—C11—H11C | 109.5 |
| C5—C6—H6 | 119.3 | H11B—C11—H11C | 109.5 |
| C1—C6—H6 | 119.3 | ||
| O1—S1—N1—C1 | 14.2 (2) | C10—C2—C3—C4 | −179.2 (2) |
| O2—S1—N1—C1 | 144.26 (18) | C2—C3—C4—C5 | −1.2 (4) |
| C9—S1—N1—C1 | −101.8 (2) | C3—C4—C5—C6 | 0.4 (5) |
| O1—S1—N1—C7 | −164.23 (18) | C4—C5—C6—C1 | 1.0 (5) |
| O2—S1—N1—C7 | −34.2 (2) | C2—C1—C6—C5 | −1.7 (4) |
| C9—S1—N1—C7 | 79.8 (2) | N1—C1—C6—C5 | −178.6 (3) |
| C7—N1—C1—C6 | 104.8 (3) | C1—N1—C7—C8 | −78.2 (3) |
| S1—N1—C1—C6 | −73.6 (3) | S1—N1—C7—C8 | 100.3 (2) |
| C7—N1—C1—C2 | −72.0 (3) | C11—O4—C10—O3 | 2.3 (4) |
| S1—N1—C1—C2 | 109.6 (2) | C11—O4—C10—C2 | −176.2 (2) |
| C6—C1—C2—C3 | 0.9 (3) | C3—C2—C10—O3 | 170.1 (3) |
| N1—C1—C2—C3 | 177.6 (2) | C1—C2—C10—O3 | −9.6 (4) |
| C6—C1—C2—C10 | −179.4 (2) | C3—C2—C10—O4 | −11.5 (3) |
| N1—C1—C2—C10 | −2.7 (3) | C1—C2—C10—O4 | 168.9 (2) |
| C1—C2—C3—C4 | 0.5 (4) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C9—H9B···O1i | 0.96 | 2.50 | 3.372 (5) | 151 |
| C7—H7B···O3 | 0.97 | 2.57 | 3.044 (4) | 110 |
| C9—H9C···O3 | 0.96 | 2.59 | 3.152 (4) | 117 |
Symmetry codes: (i) −x, −y, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2412).
References
- Bruker (2007). APEX2 (Version 1.27), SAINT (Version 7.12a) and SADABS (Version 2004/I). Bruker AXS Inc. Madison, Wisconsion, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Hanson, J. R. & Hitchcook, P. B. (2004). J. Chem. Res (M)., pp. 642–648.
- Lombardino, J. G. (1972). J. Heterocyclic Chem.9, 315–317.
- Mookherjee, B. D., Trenkle, R. W., Calderone, N. & Sands, K. P. (1989). US Patent 4 879 271.
- Reissenweber, G. & Mangold, D. (1982). US Patent 4 310 677.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siddiqui, W. A., Ahmad, S., Khan, I. U. & Malik, A. (2006). J. Chem. Soc. Pak.28, 583–589.
- Siddiqui, W. A., Ahmad, S., Khan, I. U., Siddiqui, H. L. & Ahmad, V. U. (2007a). J. Chem. Soc. Pak.29, 44–47.
- Siddiqui, W. A., Ahmad, S., Khan, I. U., Siddiqui, H. L. & Weaver, G. W. (2007b). Synth. Commun.37, 767–773.
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Tadashi, T., Jeffrey, C. G. & James, C. P. (1982). J. Biol. Chem.257, 5085–5091.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680800007X/hk2412sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680800007X/hk2412Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report