

Abstract
Early in infection, human immunodeficiency virus type 1 (HIV-1) generally uses the CCR5 chemokine receptor (along with CD4) for cellular entry. In many HIV-1-infected individuals, viral genotypic changes arise that allow the virus to use CXCR4 (either in addition to CCR5 or alone) as an entry coreceptor. This switch has been associated with an acceleration of both CD3+ T-cell decline and progression to AIDS. While it is well known that the V3 loop of gp120 largely determines coreceptor usage and that positively charged residues in V3 play an important role, the process of genetic change in V3 leading to altered coreceptor usage is not well understood. Further, the methods for biological phenotyping of virus for research or clinical purposes are laborious, depend on sample availability, and present biosafety concerns, so reliable methods for sequence-based “virtual phenotyping” are desirable. We introduce a simple bioinformatic method of scoring V3 amino acid sequences that reliably predicts CXCR4 usage (sensitivity, 84%; specificity, 96%). This score (as determined on the basis of position-specific scoring matrices [PSSM]) can be interpreted as revealing a propensity to use CXCR4 as follows: known R5 viruses had low scores, R5X4 viruses had intermediate scores, and X4 viruses had high scores. Application of the PSSM scoring method to reconstructed virus phylogenies of 11 longitudinally sampled individuals revealed that the development of X4 viruses was generally gradual and involved the accumulation of multiple amino acid changes in V3. We found that X4 viruses were lost in two ways: by the dying off of an established X4 lineage or by mutation back to low-scoring V3 loops.
Early studies of the biological properties of human immunodeficiency virus type 1 (HIV-1) found that virus isolates could be placed into as few as two phenotypic categories (defined in vitro as either non-syncytium-inducing [NSI] or syncytium-inducing [SI]) in certain CD4+ T-cell lines. These phenotypes were often found to be associated with differences in growth properties and cytopathicity on peripheral blood mononuclear cells (PBMC) (1, 14, 46) and in cellular host range (3, 48). Ultimately, the difference between the NSI and SI phenotypes was shown to be due largely to the differential use of chemokine receptors as coreceptors for viral entry: NSI viruses predominantly use CCR5, while SI viruses can use CCR5 and CXCR4 or CXCR4 exclusively (2, 29, 31, 52, 54). Results determined on the basis of SI phenotype and/or coreceptor usage typing showed that although HIV-1 present at primary infections used the CCR5 coreceptor (R5 virus) ∼90% of the time (63, 67, 68), a substantial proportion of individuals eventually developed virus that used the CXCR4 coreceptor (X4 virus). These X4/SI viruses are associated with accelerated CD4 decline and more rapid progression of HIV-1 disease (8, 28, 33, 43, 47). Little is known about the mechanisms by which these viruses come to predominate among the HIV-1 strains present in an infected person. For example, it is not known whether X4 emergence is a primary pathogenic event or is secondary to some other event, i.e., whether the virus itself causes accelerated disease progression or whether another event causes the acceleration and perhaps also leads to X4 outgrowth. Another important unanswered question is whether X4 viruses arise multiple times during the course of disease and, if so, why they do not become dominant whenever they emerge. There is also uncertainty about the frequency with which phenotype transition occurs. Phenotypic studies suggest that 50 to 60% of progressing subjects acquire X4/SI virus (26, 57, 58), but the results of a detailed longitudinal genotypic study have indicated the occurrence, sometimes transient, of at least one of four X4-associated mutations in nine of nine individuals (50).
Coreceptor usage of a particular virus is established by functional assays (growth on MT2 cells [28] or infection of indicator cell lines [64]). These assays are limited, however, in that results are generally reported only as positive or negative and provide no insight into the sequence of mutations responsible for the phenotype switch—information which may further clarify the role of X4 viruses in pathogenesis, as we discuss below. Certain mutations, particularly in the V3 loop of env (5-7, 15, 16, 22, 23, 51), are strongly associated with syncytium induction and CXCR4 usage; in particular, basic amino acids at V3 positions 11 and 25 (amino acid coordinates 306 and 322 [GenBank accession no. K03455] in standard reference HXB2) very frequently distinguish primary X4 from R5 viruses (9, 15, 16, 21, 42, 66), with positions 24 and 27 implicated in some cases (10, 37). However, while evolutionary studies of the R5-X4 transition have been undertaken (30, 38, 61, 62), the actual mutational pathway or pathways by which R5 viruses that establish infections in vivo evolve into X4 viruses (i.e., the specific evolutionary sequence of mutations required) has been largely unexplored. It is not clear whether the appearance of basic amino acids at sites 11 and 25 is sufficient or even necessary in most cases in vivo to lead to the outgrowth of X4 virus or whether, instead, a more gradual process of mutation accumulation takes place. If mutant accumulation does occur in a more gradual process, this may provide an opportunity for both early detection and arrest of X4 development. Viruses that can use both coreceptors (R5X4 viruses) are known to arise around the time of R5-to-X4 transition (2, 8, 38, 61), but their evolutionary role in that transition is not certain. The answers to questions of in vivo evolution cannot be approached by phenotypic assays alone, and analyses based on the appearance of positively charged mutations at V3 sites 11 and 25 have led to incomplete and sometimes ambiguous conclusions regarding the transition process (35).
The V3 loop is highly variable within and between individuals, and bioinformatic approaches suggest that many changes not yet examined virologically are likely to influence coreceptor usage (21, 24, 42). To gain a broader understanding of the mutations that contribute to X4 phenotype and of the temporal sequence in which they occur, we used position-specific scoring matrices (PSSM) (19, 20) to analyze V3 sequences. PSSM are used to detect nonrandom distributions of amino acids at adjacent sites associated with empirically determined groupings of sequences. They are frequently used to search DNA or protein sequences for particular motifs, e.g., transcriptional regulatory sites (55), coiled-coil domains (32), major histocompatibility complex class I binding sites (41), and others. A PSSM uses background genetic variation as a baseline comparison, or “null model,” to facilitate comparison of the residues of a sequence fragment to those of a group of aligned sequences known to have the desired property. The comparison leads to a score that can be interpreted as indicating the likelihood that the sequence fragment has the property of interest. In our study, the empirical groupings consist of V3 loop sequences associated with X4 (or SI) virus and R5 (or NSI) viruses. Using the PSSM as described below, a sequence can be assigned a score: the higher the score, the more closely the sequence resembles those of known X4 viruses.
We used the PSSM score for two purposes. First, we developed a PSSM-based phenotype predictor usable for all V3 sequences. We explored the statistical properties of this predictor and showed that it outperforms simple methods that categorize sequences on the basis of the presence of basic amino acids at sites 11 or 25. We validated the predictor with two sets of V3 sequences from phenotyped viruses different from those used to produce the PSSM matrix. Second, we showed that the score can serve as a measure of the transition from R5 to X4 phenotype.
Since the PSSM score can act as a continuous indicator of X4 evolution, we used it to identify common temporal patterns among 11 serially sampled individuals. By scoring reconstructed ancestors of the sampled virus for each subject, we demonstrated that the progression from low-scoring (R5-like) to high-scoring (X4-like) viruses was generally gradual but that the loss of putative X4 virus at the later stages of infection can occur in two different ways.
MATERIALS AND METHODS
PSSM.
A “training set” of V3 amino acid sequences from viruses of known phenotype was used to generate a matrix of likelihood ratio scores for each site in the sequence. The site-specific scores reflect the difference in abundance of a particular amino acid at a particular site in the X4 or SI group of sequences compared to that seen with the R5 or NSI group of sequences. To score a given V3 sequence, the log likelihood ratio was calculated for each site; then, the ratios for all sites were added to obtain the final score. In general, the higher the score, the more similar the given V3 sequence is to an average actual X4 sequence.
Let fij(X4) be the frequency of amino acid i (= 1 to 20) at V3 site j in a set of known X4 sequences and fij(R5) be the corresponding quantity in a set of known R5 sequences. Formally, then, the PSSM M = (mij) is defined by
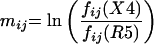 |
(1) |
and the PSSM score z for a V3 loop sequence s is given by
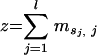 |
(2) |
where sj is the jth amino acid of s and l is the length of the sequence in amino acids.
Note that in the above formulation, likelihood ratios for amino acids i that never appear at site j in the R5 data set are undefined. To correct for this in practice, we use a standard pseudo-count procedure in the matrices which amounts to initializing every amino acid at one count at every site and then counting actual representations of each amino acid in the data set (19). That is, we replace fij as defined above with
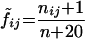 |
(3) |
where nij is the number of times amino acid i appears at site j in the data set and n is the total number of sequences in the data set.
This method requires that all sequences be of identical lengths. Most training sequences were 35 amino acids long. There were 44 length variants (ranging from 34 to 38 amino acids) in the training data. Gaps were inserted as necessary to align homologous residues; insertions were removed and only the remaining amino acids were scored. Gaps were considered to contribute a value of 0 to the PSSM score of a sequence. The performance of the prediction method on sequences with length variation is examined below.
Data sets.
To develop the motif scoring matrices, we used HIV-1 clade B V3 loop sequences obtained from biologically cloned viruses whose MT2 tropism (SI or NSI) or coreceptor usage (CCR5, CXCR4, or dual usage) had been determined. We used the sequence sets described by Resch et al. (42); these are referred to below as the SI/NSI set (for which only MT2 tropism was assayed) and the X4/R5 set (for which coreceptor usage was assayed). Dual coreceptor usage was also noted for sequences in the X4/R5 set. The SI/NSI set contained 70 SI and 187 NSI sequences from 107 subjects; the X4/R5 set contained 17 X4, 168 R5, and 28 dual-tropic sequences from 177 subjects. Unless specifically mentioned otherwise below, the dual-usage viruses were grouped with the X4 class.
To test the validity of the phenotype prediction method developed using the scoring system, we analyzed a separate set of 175 V3 sequences of known phenotype. This set consisted of biologically cloned viral isolates (from four men who have sex with men) collected in the Amsterdam Cohort Study (ACS) (12) and obtained over multiple timepoints before, during, and after an observed coreceptor usage switch (61). Virus was sampled every 3 months for between 4 and 6 years after seroconversion.
We then used the PSSM to reanalyze sequence data obtained at multiple timepoints from 11 subjects in the Multicenter AIDS Cohort Study (MACS) (25), 9 subjects from the study of Shankarappa et al. (50) and 2 individuals with newly obtained sequence data. All 11 were men who have sex with men and had enrolled prior to HIV infection in an ongoing longitudinal study of HIV disease. Sampling occurred every 6 months over the entire course of infection, extending 6 to 14 years.
Phenotype prediction.
The designing of a phenotype predictor on the basis of a PSSM score requires two points to be considered. First, given a PSSM generated from known sequences, one must choose a threshold score above which the virus is predicted to use CXCR4 (and possibly CCR5) and below which it is predicted to use CCR5 exclusively. The choice of this threshold or cutoff value ideally will jointly maximize the specificity and sensitivity of the predictor.
Second, the issue of choosing a cutoff value score is complicated by the fact that the scoring matrix is dependent on a sample of all possible V3 loops and their associated phenotypes. If the loops in this sample were to differ in size or in sequence, the matrix values would also differ and so would the score assigned to any given sequence. To estimate the extent of this sampling variability, we used a bootstrapping procedure (34) in which many matrices were calculated on the basis of subsamples of the original training data set. These matrices were used to score each sequence in the set, generating an estimated distribution of scores for each sequence. This variability in the scores can be taken into account in the design of a prediction method and can be used to perform statistical tests of the method's effectiveness. The cross-validation analyses (see below) also provide a measure of sampling variability.
Choice of cutoff value.
For each matrix calculated, we identified the cutoff score that when applied to the training data set maximized the association coefficient; we refer to this score as the optimal cutoff value for the given matrix. The coefficient is given by
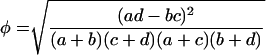 |
(4) |
where a represents the number of true positives (i.e., X4 or SI variant scores higher than the cutoff value), b represents false positives (R5 or NSI scores higher than the cutoff value), c represents false negatives (X4 or SI scores lower than the cutoff value), and d represents true negatives (R5 or NSI scores lower than the cutoff value). This quantity runs from zero (when the PSSM score is indiscriminate with respect to phenotype) to unity (when the score perfectly divides the sequences into phenotype classes).
Analysis of sampling variability.
To estimate the variability in scores and optimal cutoff values induced by the effects of sequence sampling on the PSSM matrix, we performed bootstrapping as follows. For a given sample size, X4 and R5 sequences were randomly drawn with replacement from the data set under analysis in proportions equal to those observed for the entire data set. Using this sample, a PSSM was constructed as described above. With this matrix, a score was calculated for each sequence in the training data set. For this set of sequence scores, an optimal cutoff value was calculated. Sampling, matrix construction, and scoring were performed 100 times, generating a distribution of scores, optimal cutoff values, and association coefficients for each sample size. To examine the effect of increasing numbers of samples on the efficiency of the predictors, we adjusted the sample sizes from 25 to the size of the data sets in increments of 25.
We analyzed PSSM constructed using both the SI/NSI set and the X4/R5 set. Matrices were applied to the entire training set. Previous analyses of these data sets (21, 42) used randomly selected subsets of data in which (at most) one sequence from any single individual for each phenotype was allowed. This was done to reduce spurious correlations between sequence and phenotype induced by the evolutionary dependence of sequences within individuals. However, because the sequences were numerous and also were chosen such that each V3 loop amino acid sequence was unique, we reasoned that using the entire set of sequences to form the scoring matrix would be justified and would lead to better R5/X4 discrimination. That is, since every sequence was unique, some independent evolution occurred in those sequences coming from single subjects, and since the sequences were numerous, it is likely that mutations not influencing phenotype would arise at approximately the same rates for either phenotype and consequently have no influence (on average) on the overall score. One possible caveat is that if a few patients were to contribute many sequences, the evolutionary dependence among those sequences would lead to artifactually high levels of predictor performance. This issue is partially mitigated by the uniqueness of all the V3 sequences; we also note that no patient accounted for more that 4.6% of the sequences in the SI/NSI set or more than 2.9% of those in the X4/R5 data set. We tested the hypothesis that the use of the entire data set would lead to better performance by using the bootstrap method described above, generating bootstrapped sequence sets derived either from unique subjects or from the entire set and comparing the resulting distributions of the association coefficient as a measure of predictor efficiency. We also performed a similar comparison using cross-validation analysis (see below).
Cross-validation.
The bootstrap analysis gives an idea of the effect of sequence sampling on the variability in the performance of the PSSM. However, since the matrices in this analysis are reapplied to the set of sequences that was sampled to derive those matrices, the performance measures are likely to be inflated. Also, the positive predictive value (PPV; i.e., the fraction of predicted CXCR4-using virus that actually use CXCR4, given the X4 prevalence in the infected population) of the test, as applied to the training set, may be inflated relative to that expected when the test is applied to new clinical sequences, since the prevalence of X4 virus in infected individuals at large is probably lower than the ∼20% representation in the training set. To address these issues, we performed a cross-validation analysis.
N-fold cross-validation (for examples in an HIV sequence analysis context, see references 40 and 45) of the PSSM method was performed as follows. For each iterate, the X4R5 data set was randomly partitioned into N subsets. The choice of N is discussed below. For each subset i, the remaining subsets 1,…, i-1, i+1,…, N were pooled and used to develop a PSSM. This PSSM was used to score the sequences in subset i, and the quality measures (association coefficient, PPV, sensitivity, and specificity) associated with the optimal cutoff value for that matrix and subset were recorded. Thus, N sets of quality measures were generated for each partition. For each iterate, the average of the values of each measure over the N values was reported. 100 iterations yielded a distribution of average quality measures for each set of parameters. To provide a comparison with the 11/25 rule, we generated distributions of average quality measures by partitioning the data set as described above, applying the 11/25 rule to each subset and recording the averages for 100 partitions. We also use this analysis to examine the modified charge rule (as suggested by Hoffman et al.) (21), in which R or K at position 11, or K at position 25, predicts X4 virus. The distributions of quality measures were compared using the Kruskal-Wallis test.
For a data set of given size, a small value of N (close to 2) provides the largest validation set but the smallest training set and a large value of N (approaching the size of the data set) focuses a large training set on a smaller validation sample. In the former case, prediction reliability may suffer; in the latter, small samples may increase variability in the quality scores. In the investigation of sensitivity and specificity with the entire data set, we performed cross-validation studies for a range of N values and found that variability was minimized and that quality measures peaked around N = 10 (data not shown). We report N = 10 results here.
For the cross-validation study of X4 PPV, we wished to examine PPV over a range of prevalence fractions that we believe spans the actual X4 prevalence in the infected population. We chose the values for N so that at the lowest prevalence, the validation set would consist of a single X4 (or R5X4) V3 loop and a number of R5 V3 loops. For each iterate, the X4/R5X4 and R5 sets were each randomly divided into N groups. To guarantee a constant prevalence level, the validation set for each group was created by sampling a given number of X4 and R5 sequences with replacement from the ith group and the remaining groups were pooled to create the scoring matrix. To examine a minimum X4 prevalence of ∼2% according to this scheme, we required a partition size of N = 4 for the X4/R5 data and N = 5 for the SI/NSI data.
Design of the composite predictor.
Because sampling leads to variability in scores for a given sequence, the optimal cutoff value for a matrix used to define the predictor is also inherently variable. Sequences that have intermediate scores are essentially randomly assigned an X4 or an R5 prediction according to the optimal cutoff value calculated for a particular training set. To reduce this ambiguity, we designed a predictor as follows. First, on the basis of performance considerations (described in Results), we chose to score sequences using the matrix calculated for the entire X4/R5 data set. We assigned an X4 prediction to any score higher than the 95th-percentile of optimal cutoff values, i.e., any sequence that scored higher than the optimal cutoff values for 95 of 100 bootstrap-generated matrices. Similarly, we assigned an R5 prediction to any score lower than the fifth percentile of optimal cutoff values. These two values are referred to as the X4 and R5 cutoff values, respectively. The unassigned, intermediate scores were then assigned a prediction based solely on whether they possessed basic amino acid residues at either site 11 or site 25. We refer to this predictor as the composite predictor.
Comparison with charge-based methods.
The 11/25 method, in which a sequence is predicted to be X4 when it harbors arginine or lysine at V3 site 11 or 25, is a widely used method for sequence-based prediction of phenotype (15, 21, 39, 50). Hoffman et al. (21) also suggested the use of a modified 11/25 method in which arginine at position 25 is disregarded in prediction. We compared PSSM with an optimal cutoff value to these methods using cross-validation and evaluated the composite predictor by applying all methods to the SI/NSI and ACS data sets and calculating the reliability (i.e., the proportion of correctly predicted sequences out of all predicted to be X4/SI), specificity (the proportion of actual R5/NSI correctly predicted), and sensitivity (the proportion of actual X4/SI correctly predicted) of each method. The cross-validation procedure allowed us to compare the methods statistically.
External validation.
To examine the performance of the method with data not used in the matrix formulation, we applied the composite predictor to the ACS data set. We report reliability, sensitivity, and specificity for both the charge-based and X4/R5-based composite methods. We also report quality measures for the composite method as applied to the SI/NSI data set. Although the two classifications do not exactly overlap, this is one way to uniformly compare the methods (since the charge-based methods do not distinguish between the two classifications).
In silico mutagenesis of training sequences.
To examine whether changes at sites 11 and 25 alone could substantially shift the PSSM score distribution of our sample population and thus lead to appreciable changes in the predicted phenotypes, we sequentially and singly changed sites 11 and 25 to arginine and lysine in the R5/NSI data, changed these sites to consensus (R5/NSI) residues in the X4/SI data, and recalculated the score distributions.
Comparison of score distributions between coreceptor usage classes.
We calculated the score means and distributions for separate R5, R5X4, and X4 groups of V3 sequences in the X4/R5 and ACS data sets. Statistical comparisons of the distributions were performed using the Tukey-Kramer method as implemented in the program JMP (SAS, Cary, N.C.).
Viral gene sequencing.
Viral populations from subjects 4 and 10 (44) were sampled and sequenced using previously described protocols (50). For subject 8 from the Shankarrappa et al. (50) study, we included sequences from nine biologically cloned and phenotyped virus isolates sampled from PBMC at two timepoints: 0.29 years after seroconversion, at which time only NSI or R5 virus had been observed, and 5.45 years after seroconversion, the time of the first visit at which SI/X4 virus had been observed. Isolation and phenotyping was performed as described elsewhere (62).
Phylogenetic reconstruction and ancestral V3 loop estimation.
We used data from subjects 1, 2, 3, 5, 6, 7, 8, 9, and 11 (previously obtained by Shankarappa et al.) (50) and additional sequences we derived from subjects 4 and 10 and from biologically cloned viral isolates from subject 8 to generate phylogenetic trees of env C2V5 sequences. Sequence editing and contig assembly were performed using Sequencher software, version 3 (Gene Codes Corporation, Inc., Ann Arbor, Mich.). Sequences were aligned using CLUSTALW (59) and then manually edited. Phylogenetic analyses were performed using PAUP*, version 4.0b10 (56). Phylogenetic trees were inferred by first estimating a neighbor-joining tree using maximum likelihood distances and then swapping branches on this tree under maximum likelihood using the subtree pruning and regrafting algorithm for subjects 1, 2, 3, 5, 6, 7, 8, 9, and 11 and the nearest-neighbor interchange algorithm for subjects 4 and 10 (56). Models of sequence evolution were estimated under maximum likelihood using the general time-reversible model of substitution with unequal base frequencies and among-site rate heterogeneity (56). We used these trees as a basis for reconstructing putative ancestral V3 loop sequences at each internal node. The PSSM score for each putative ancestor was calculated. To visualize the evolutionary change of score within the phylogeny, we used routines written in PERL and Mathematica (Wolfram Research, Champaign, Ill.) to apply colors to branches and nodes that reflected the scores of the predicted ancestors they represented.
Supplementary material.
Supplementary figures accompanying this work can be accessed at the URL http://ubik.microbiol.washington.edu/HIV/Jensen2003.
Nucleotide sequence accession numbers.
The sequences discussedin this work can be found in GenBank under accession numbers AF137629 to AF138163, AF138166 to AF138263, AF138305 to AF138703, AF204402 to AF204670, AY348333 to AY348528, AY348532 to AY348544, and AY449806 to AY450257.
RESULTS
Phenotype prediction.
We used bootstrap estimates of sampling variability in predictor performance as well as cross-validation analysis to answer the following questions. (i) Would a larger number of training sequences be likely to substantially improve performance in predicting R5/NSI and X4/SI phenotype? (ii) Would the inclusion of multiple sequences from the same patient improve performance? (iii) What cutoff values should be used for the X4 and R5 cutoff values in the composite predictor (see Materials and Methods)? (iv) How does PSSM performance compare to that of charge rules, particularly in light of the relative rarity of X4 viruses?
Figure 1 shows the results of the use of the association coefficient to summarize the bootstrap variability analysis for PSSM derived from the X4/R5 training data set and applied to X4/R5 data (Fig. 1A) and SI/NSI data (Fig. 1B) as a measure of prediction performance. Similar general patterns were seen when PSSM derived from the SI/NSI training data set were used (see supplementary Fig. S1). With respect to whether increasing the number of training sequences would improve performance (question i), we found that the response of the mean association coefficient to increasing sample sizes became relatively flat after the sample size reached about 100 sequences for the SI/NSI data set, though the variability continued to decrease as more sequences were added (Fig. 1B). The mean association continued to climb as sequences for the R5/X4 data set were added, but the variability remained constant and large. The results depicted in the graphs suggest that (at least with the present X4:R5 proportion) substantial improvement in the performance of the PSSM method would require the addition of a relatively large number of sequences (>100) to the training sets. The addition of X4 sequences alone (to bring their numbers in line with the R5 sequences) might lead to greater improvement in the accuracy of results than that suggested by the foregoing analysis (see below).
FIG. 1.

Bootstrap analysis of R5/X4 data set PSSM. Error bars delineate the 5th and 95th percentiles of the bootstrapped distribution for the association coefficient. The numbers of sequences sampled to produce matrices are indicated on the X axis. Diamonds represent results for sequences guaranteed to be sampled from a different subject; squares represent results for sequences sampled from all data. (A) Matrices produced by sampling X4/R5 data (X4/R5 matrices) applied to X4/R5 data; (B) X4/R5 matrices applied to SI/NSI data; (C) optimal cutoff value distribution for X4/R5 matrices (generated by sampling the entire X4/R5 data set [213 sequences]) applied to the combined data set. The boxes under the X axis indicate quartiles; error bars indicate 5th and 95th percentiles, chosen as the R5 and X4 cutoff values, respectively, for the composite predictor.
With respect to whether we are justified in including multiple sequences from the same individual (question ii), we found that the mean association was slightly better when all the sequences are used than when only single sequences of either phenotype from any given individual were used, although the discrimination ability were not significantly different in either case (Fig. 1A and B). Cross-validation also indicates that specificity and sensitivity are significantly higher, and variation is reduced, when all the data are included (Fig. 2; for the difference between the results for all data versus that for unique distributions, p < 0.0005 [Kruskal-Wallis test]). Recall that in this procedure, the portion of the data set being evaluated is not used to train the prediction method. Figure 1C addresses the question of appropriate cutoff values (question iii). For the PSSM derived from the entire X4/R5 data set, the 95th-percentile optimal cutoff value (X4 cutoff value) was −2.88 and the 5th-percentile optimal cutoff value (R5 cutoff value) was −6.96. That is, the value for any V3 scoring higher than the X4 cutoff value would exceed the optimal cutoff value for 95% of PSSM matrices generated by sampling and the value for any V3 scoring lower than the R5 cutoff value would be lower than the optimal cutoff value for 95% of PSSM matrices. In this sense, such V3 sequences have significantly high or low scores (relatively independent of the sequence sample used to create the PSSM) and should be predicted X4 or R5, respectively.
FIG. 2.

Cross-validation comparison (using the X4/R5 data set) of PSSM and charge-based methods. The partition size was 10 (see text for a description of the method). All data, all sequences employed in the PSSM analysis; Unique, single X4 and R5 sequences chosen from patients in PSSM analysis; 11/25, 11/25 method; 11/25 mod, modified charge method; Sens, sensitivity (with respect to X4 prediction); Spec, specificity. Error bars are at points 1.5 times the interquartile range from the box; outliers are shown as open circles.
The cross-validation results for the present data (Fig. 2) suggest how the PSSM method with optimal cutoff value and the charge-based method (including the 11/25 charge rule and the modified 11/25 rule [in which K only at site 25 predicts X4]) compare as general approaches. When all data were used in unaltered form (∼20% R5X4 or X4, ∼80% R5), the levels of both median specificity and sensitivity were significantly higher for the PSSM method than for the standard rule (p ≪ 10−5). The modified 11/25 rule method was significantly less sensitive than the two other methods but was significantly more specific. The medians and ranges for these values (sensitivity median, 0.51; sensitivity range, 0.50 to 0.54; specificity median, 0.95; specificity range, 0.95 to 0.95) were comparable to those found using a similar analysis of Hoffman et al. (21) (sensitivity median, 0.49; sensitivity range, 0.45 to 0.52; specificity median, 0.95; specificity range, 0.95 to 0.95).
Effects of varying X4 prevalence.
We chose the range of X4 prevalence (proportion of infected individuals having X4 virus) on the basis of the following observations. The standing natural prevalence (in the absence of therapy) can be crudely estimated with a simple model: suppose that HIV prevalence in a population is stable; this approximates the situation in Europe and the United States. Then, according to the model presented in the Appendix, the equilibrium X4 prevalence is β/(β + δx) where β is the rate of conversion from R5 to X4 virus per R5 individual per year and δx is the death rate of individuals harboring X4 virus per year. According to Koot et al. (27), if SI virus is considered equivalent to X4 virus, β is 4.6%/year for individuals with CD4+ counts of >500/μl and 8.0%/year for those with counts of <500/μl. The diagnosed rate of AIDS in the X4 category is estimated to be 38.8%/year (36). If we approximated the death rate with the AIDS rate, since X4 lineages harbored by very ill individuals are likely to be dead in the epidemiological sense (see comment in Appendix) the steady-state natural prevalence of X4 would be between 10.6 and 17.1%. These estimates suggest the upper limit of prevalence that would be observed in cohorts of patients who are not on therapy and have declining CD4+ T-cell counts. Harrigan et al. have recently used the PSSM method described here to analyze baseline V3 consensus sequences (P. R. Harrigan, W. W. Y. Dong, B. Yip, Z. L. Brumme, B. Wynhovenm, N. Hoffman, R. Swanstrom, T. Mo, M. A. Jensen, J. I. Mullins, R. S. Hogg, and J. Montaner, Abstr. 2nd Int. AIDS Soc. Conf. HIV Pathogenesis and Treatment, Paris, France, abstr. 143, 2003) of 1,107 persons starting suppressive antiretroviral therapy (interquartile CD4+ counts, 130 to 420). Of these persons, 111 were predicted by the composite method to harbor X4 virus; thus, a rough estimate of actual prevalence would be 111 × 89% reliability/1,107 = 8.9%. The result of this calculation corroborates the lower of the estimates given above. This group represents the population most relevant to a possible diagnostic test.
On the basis of these considerations, we examined PSSM predictive value for X4 prevalences from ∼2 to ∼10%. Cross-validation results for PPV are shown in Fig. 3. The variance is large and suggests again that more sequence and phenotype data might improve the method. However, the PSSM distributions were significantly higher than those from the charge-based methods at prevalences approaching the natural prevalence for both the X4/R5 and SI/NSI data sets (Kruskal-Wallis P < 0.001; 7.4% prevalence and above in the X4/R5 set and 4.0% and above in SI/NSI set). PSSM did particularly well at predicting SI virus at low levels of prevalence. The improvement in X4 prediction was apparently due to an increase in overall specificity as more X4 sequences are added, while improvement in SI prediction was due to increasing sensitivity (supplementary Fig. S2).
FIG. 3.

Cross-validation comparison (with various X4 or SI prevalence values) of PSSM and charge-based methods. (A) X4/R5 data set; (B) SI/NSI data set. PPV, PPV with respect to X4; Prevalence, percent X4 sequences in the validation subsets; 11/25, 11/25 method; 11/25 mod, modified charge method. Error bars are at points 1.5 times the interquartile range from the box; outliers are shown as open circles.
These results also suggest that the most efficient way to improve the PSSM, in terms of expanding the training set, would be to add more X4 sequences.
PSSM for subsequent analyses.
Since using all the sequences improved the statistical performance of the predictor, we chose to use all sequences in a training set to derive the PSSM. On the basis of the optimal cutoff value, the X4/R5-derived matrix gave 83% reliability (PPV given the prevalence in the data set in unaltered form) with the X4/R5 data and 89% reliability with the SI/NSI data and the SI/NSI-derived matrix gave 96% reliability with the SI/NSI data but only 61% reliability with the X4/R5 data. Sensitivity and specificity results were similar. Since this matrix performed better on the alternative data set and was based entirely on sequences of biological clones of experimentally determined phenotype rather than on samples of bulk viral isolates, we used the X4/R5 matrix as the basis of the composite predictor in the following analyses. Further, scores of dual-usage (R5X4) viruses can be considered separately from those of X4 and R5 viruses (both matrices appear in supplementary Fig. S1). X4 and R5 cutoff values for the composite predictor are shown in Fig. 1C. (The composite predictor uses the 11/25 predictor for sequences which score between the X4 and R5 cutoff values [see Materials and Methods]). The frequency distribution of scores for the entire training set is shown in Fig. 4.
FIG. 4.

Score distributions for all training sequences. Y axis, frequency within coreceptor usage class; X4/dual/SI, sequences associated with CXCR4 usage or syncytium induction; R5/NSI, sequences associated with pure CXCR5 usage or inability to form syncytia; 11/25+, sequences containing basic residues at site 11 and/or 25; 11/25−, sequences not containing basic residues at these sites. Vertical lines indicate R5 and X4 cutoff values as described in the text. Note that this is not a typical frequency histogram but is a superposition of the R5/NSI and X4/SI frequency histograms; the total area of the bars sums to 2. (The X4/SI subset is too small relative to the R5/NSI subset to be visualized easily as part of an ordinary histogram.) The solid fractions of mixed bars indicate sequences correctly predicted by the canonical predictor; the fractions shaded light and dark gray were incorrectly predicted.
We compared the performance of the composite X4/R5-based predictor to that of the 11/25 predictor for the SI/NSI training set (see Materials and Methods) and the ACS data (Table 1). The composite predictor (determined on the basis of the X4/R5 data set) had intermediate sensitivity (84%; 11 samples miscalled SI out of 70 samples) and comparable specificity (96%; 7 miscalled NSI out of 187) to those seen with the 11/25 rule for the SI/NSI test set. The reliability (89%) for this set was lower than that of the other two methods. This does not contradict the cross-validation results, as those analyses considered PSSM performance within data sets of like virological data. We do not know whether the miscalls were due to incorrect classification by the method or to the possibility that SI phenotype of the given virus did not match its coreceptor usage. For example, bulk isolates were used in some of the SI/NSI assays. Such an isolate might contain mostly R5 virus and yield an R5 V3 consensus sequence, but a small amount of X4 virus in the isolate would infect the MT2 cell line and lead to a positive SI assay. Length variants in the complete training set were also considered in a separate analysis, since in the simple way we handled gaps and insertions (i.e., gaps contributed a value of 0 to the score), some information contributed to the score by the PSSM was lost at the gaps. However, the results in Table 1 show that the use of this method allowed the classification of these length variants as well as that of the typical sequence. The reliability was better for this subset than for the entire set, which suggests that deviation from the typical 35-amino-acid length is frequently associated with CXCR4 usage. Both prediction methods performed well with the ACS validation set.
TABLE 1.
Performance of charged-based and composite predictors
| Data set | n (x + r)b | Results (%) by methoda
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 11/25
|
Modified 11/25
|
Composite
|
||||||||
| Sens | Spec | Rel | Sens | Spec | Rel | Sens | Spec | Rel | ||
| SI/NSI | 257 (70 + 187) | 90 | 96 | 90 | 76 | 98 | 95 | 84 | 96 | 89 |
| Length variants alone | 44 (25 + 19) | 72 | 100 | 100 | 72 | 100 | 100 | 76 | 95 | 95 |
| ACS | 175 (81 + 94) | 81 | 99 | 99 | 81 | 99 | 99 | 89 | 100 | 100 |
For each data set and predictor, the specificity (Spec; proportion of true X4/R5X4/SI sequences correctly predicted by method), sensitivity (Sens; proportion of true R5/NSI sequences predicted), and reliability (Rel; proportion of predicted X4/R5X4/SI sequences actually having that phenotype) are presented. Length variants (V3 sequences not having 35 amino acids) from the combined training set are also analyzed separately.
Results represent the total number (n) of sequences (number of CXCR4-using or SI [x] and number of pure CCR5-using or NSI [r] sequences).
Basic residues at sites 11 and 25 are strongly associated with an X4/SI phenotype in subject-derived virus, as demonstrated by the good performance of the charge-based predictors described above. However, it does not necessarily follow that positively charged mutations at those sites induce that phenotype in all sequence backgrounds or that mutations to uncharged residues at those sites in X4 viruses cause a reversion to the R5/NSI phenotype. Figure 5A shows the impact of single-residue basic mutations at sites 11 and 25 on the distribution of PSSM scores of known R5/NSI V3 loops. Depending upon the mutation, the composite predictor assigned between 8.2 and 38.1% of mutated sequences to the X4 class; the baseline false-positive assignment was 1.7% (Fig. 5A). We also note that 25K had a stronger effect on scores than 25R, corroborating the effect noted by Hoffman et al. (21) that motivated the modified 11/25 rule. Thus, many (but not the majority) of the mutated sequences were likely to be X4 sequences as predicted as a result of a basic amino acid substitution at either of these two sites. On the other hand, single-residue reversions of 11/25 mutations to R5/NSI consensus residues in X4/SI sequences led to the loss of about half of the originally predicted X4 sequences (i.e., from 76% to approximately 50% of the sequences predicted). These results together suggest that for many V3 backgrounds, basic changes at 11 or 25 are neither necessary nor sufficient for a phenotype switch. Mutagenesis at site 11 or 25 of known R5/NSI sequences yielded no known X4/SI sequences in our data sets. Three X4/SI-11S mutants and four X4/SI-25D mutants were found in the known R5/NSI data set.
FIG. 5.

Impact of V3 loop site 11 and 25 mutations on PSSM score. The results for V3 score distributions from known R5/NSI (A) and X4/SI (B) viruses and the same sequences with substitutions at positions 11 and 25 are shown. Boxes indicate quartiles; error bars indicate 10th and 90th percentiles. Percentages indicate the proportions of sequences predicted by the composite predictor to be CXCR4 users.
If single changes at 11 or 25 do not result in a change of coreceptor usage in many backgrounds, then the virus must accumulate multiple V3 loop substitutions to shift from exclusive use of CCR5 to exclusive use of CXCR4, assuming that the scores correlated well with biological phenotype. Dual-usage (R5X4) and dual-tropic (T-cell line and macrophage-infecting) viruses are thought to represent evolutionary intermediates in this shift (18, 53). If dual-usage V3 loops tended to have intermediate PSSM scores, this would support the evolutionary-intermediate hypothesis and demonstrate that the PSSM score can serve as a continuous measure of X4 evolution.
We compared the score distributions of the different phenotype classes in the X4/R5 and ACS data sets. In the ACS (Fig. 6A), the score distribution of dual-usage virus was significantly different from and intermediate to those of pure R5 and pure X4 virus (P < 0.001, Tukey-Kramer test). The high level of significance may have been due in part to the fact that the 174 samples were obtained from four subjects, so that the sequences were not statistically independent. In the X4/R5 training set, the dual-usage distribution was bimodal; 6 of 22 dual sequences scored higher than 6.8 and the remainder scored lower than 0 (Fig. 6B). The distribution excluding the six high-scoring dual-usage results differed from the other two distributions (P < 0.01 [Tukey-Kramer test]). Thus, while there was appreciable overlap of the score distributions, dual-tropic viruses had, on average, intermediate PSSM scores.
FIG. 6.

PSSM score distributions of sequences with defined coreceptor usage. Boxes indicate quartiles; error bars indicate 10th and 90th percentiles. R5, R5X4, and X4 indicate viruses able to enter coreceptor-transfected indicator cell lines expressing CCR5 only, both CCR5 and CXCR4, or CXCR4 only. (A) ACS data, sequences from the ACS (61). (B) Low R5X4, R5X4 sequences omitting six high-scoring outliers. Differences in jittering data in the horizontal plane account for the visual differences in data between R5X4 and low R5X4; the same data are represented in each. Boxes represent interquartile ranges, with the interior line at the median; error bars are placed at the 10th and 90th percentiles.
Evolutionary reconstruction of PSSM scores and the X4 phenotype.
We generated phylogenetic reconstructions of viral sequences for each of the MACS subjects evaluated in the Shankarappa et al. study (50) and for two additional subjects (subjects 4 and 10) (44), and estimated and scored ancestral V3 loop sequences at each node in the phylogenies. Figure 7 displays three of viral phylogenies for the 11 subjects (trees for all subjects can be found in supplementary Fig. S3 through S10).
FIG. 7.



Representative phylogenetic reconstructions for subjects (Subj) 8 (A), 7 (B), and 1 (C). Colors of tip symbols indicate the years after seroconversion each sample was obtained. Colors of nodes and branches reflect PSSM scores of the reconstructed ancestors or the sample (tip) V3 sequences; cooler colors represent lower values and warmer colors represent higher values, as indicated by the scale. Various extreme scores among subjects were seen, so the color scales differ among the trees. For each tree, however, light green represents a value of −5, approximately intermediate between the R5 and X4 cutoff values. Branches are colored according to the score of the sequence of the branch's right-hand node. For subject 8 (A), filled triangle symbols on the tree represent sequences obtained from biological clones derived from the given time point. Phenotypes of these clones are indicated by the callouts. Scale bars indicate genetic distances along branches.
The subject 8 tree (Fig. 7A) places the sequences of subject-derived biological clones in the context of the original sequences. The sequences of the clones clustered with other plasma and PBMC-derived sequences obtained from samples taken at the corresponding clinic visits. The R5 clones of the later visit associated with a low-scoring cluster, while the dual-tropic clones associated with a higher-scoring cluster. The two clusters coexisted in the viral population. The scores of the dual-tropic clones were identical (−7.74). This result is within the central 50% of the distribution of dual scores shown in Fig. 6, though since the score is lower than the 5% cutoff value, they were predicted by the composite predictor to be R5. However, these sequences contained a basic mutation at site 11 and a three-residue insertion N terminal of the V3 crown. This finding, together with the results predicted on the basis of the length variants mentioned above, suggests that the 11/25 predictor is better for V3 loops longer than 35 amino acids. The fact that these clones are dual tropic suggests that the associated clade at the top of the tree is able to use CXCR4.
The subject 7 and subject 1 trees (Figs. 7B and C) exemplify two evolutionary patterns of high-scoring, putative X4 viruses. In subject 7 (and subject 3) (see supplementary Fig. S4), multiple divergent lineages coexisted over much of the infection, only one of which evolved high-scoring virus. Near the end of infection, the high-scoring lineage died out, leaving preexisting low-scoring viruses behind. For subject 1 (and for two lineages in subject 2 [supplementary Fig. S3]), high-scoring viruses evolved as an intermediate evolutionary stage. The coloration of the tree highlights the reversion of the high-scoring population to a low-scoring one caused by further mutational changes rather than lineage replacement. In the remaining individuals, high-scoring viruses either did not evolve or persisted to the end of the infection or follow-up.
Figure 8 depicts the persistence of high- and low-scoring lineages over infections in relation to disease and therapy milestones. Dual-shaded bars indicate that the sequences at that sampling time clustered into at least two monophyletic groups, one whose most recent common ancestor scored higher than −5 (the approximate midpoint between the R5 and X4 cutoff values) and another whose ancestor scored below −5. High-scoring lineages arose in most individuals and were frequently in competition with separate low-scoring lineages. They persisted at least 1 year but often were ultimately lost from the viral population. Phylogenies used in this graph are available in the supplementary material.
FIG. 8.

Coexistence of high- and low-scoring lineages. Solid black stretches indicate that sequences representing low-scoring (<−5) ancestors alone were present at the indicated times; two-pattern (solid and stippled) stretches indicate the coexistence of high (>−5)- and low-scoring lineages; stippled stretches indicate the presence of high-scoring lineages alone. The graph was devised on the basis of inferences made by inspection of reconstructed phylogenies (supplementary Fig. S3 through S10). Inverted filled triangles indicate times of first visits at which CD4+ counts were <200/μl; open triangles indicate timepoints of CD3+ T-cell inflection (accelerated decline; see reference 17); crosses indicate deaths of subjects; H indicates initiation of suppressive antiretroviral therapy. Hatched regions indicate time periods for which sequences were not available.
DISCUSSION
The role X4 viruses play in HIV pathogenesis has been elusive on several levels. Their presence clearly increases risk of disease progression, but not every individual who progresses appears to acquire this phenotype (26, 28). However, genotypic analysis has suggested that their representation had been underestimated (50) in the earlier biological studies; in particular, X4 viruses may arise but not persist. The results of the present study indicated that not all progressors develop X4 viruses predicted by PSSM matrices (8 of 11 did) but did confirm their transience in some individuals (4 of 11). There are strong genetic determinants for CXCR4 usage that evidently are not transmitted but evolve independently in a large fraction of untreated individuals. The repeatability of this phenomenon is striking in itself, as it represents repeated independent evolution to the same endpoint and suggests that the coreceptor transition might follow a common evolutionary pathway in many cases. However, no single set of mutations appears to lead to coreceptor switching in every genetic background.
We explored whether it would be more fruitful to suppose that certain mutations increase the probability that a given virus is X4, with other influences such as the larger viral genetic background also impinging on coreceptor usage. The performance of our PSSM-based method can be interpreted in this light. Empirically, the more X4-associated mutations that appear in a V3 loop the higher the score and the more likely that V3 is correctly predicted to be associated with an X4 phenotype. The association of intermediate scores with an intermediate (or at least qualitatively distinct) phenotype, dual coreceptor usage, make a model of independent accumulation of mutations leading to CXCR4 usage biologically plausible. Such a model also predicts that although certain mutations (e.g., mutations at sites 11 and/or 25) may have disproportionate influence on coreceptor usage, such mutations are not necessary for coreceptor switching (provided V3 has accumulated enough other mutations with smaller effect). Consistent with this, the data contain the results for a number of high-scoring CXCR4-using viruses that did not harbor 11/25 changes (Fig. 4). Sites 24 and 27, previously implicated as contributors to the X4 phenotype, affect the PSSM score by 0.8 and 1.1 log-odds units (50th and 80th percentiles over all V3 sites) on average, while 11 and 25 contribute 1.5 and 1.2 log-odds units (95th and 90th percentiles) on average. Finally, the propensity to use CXCR4 may not lead to the actual ability to use it in particular circumstances; conversely, certain genetic backgrounds may support CXCR4 usage in spite of an unlikely V3. The presence of variability in scores within each usage class supports this risk-based model of X4 genetics.
The mutational differences reflected in PSSM score variability may account for some of the discrepancy between the results of phenotypic and genotypic studies of the frequency of X4 development among progressing individuals. It is unlikely, however, that this accounts for the entire difference. Genotypic studies have often used basic mutations as predictors of CXCR4 usage, but our study suggests that in the absence of other X4 mutations, these mutations do not always confer a high propensity to use CXCR4 (Fig. 5). Subject 6 virus had the lowest mean scores, but this individual was nevertheless the fastest progressor in the MACS set. The results of our reanalysis indicate that he was unlikely to have harbored X4 variants, yet a previous study reported finding one X4 sequence on the basis of the presence of a basic residue at position 24 (50). This is consistent with the idea that X4 virus is not always required for progression to disease (11). Two of seven V3 sequences sampled at 5.8 years after seroconversion for subject 11 had basic residues at site 25 and were predicted by PSSM to represent X4; however, these viruses did not give rise to a persistent high-scoring lineage (supplementary Fig. S10). Subject 11 was a slow progressor (and is now being treated with highly active antiretroviral therapy) (Fig. 8). Of two individuals not evaluated in a previous study (50), subject 4 was a moderate progressor and did develop viruses predicted to be X4 and subject 10 was a nonprogressor and did not develop predicted X4 viruses.
The observation that the presence of biologically phenotyped SI virus is strongly associated with CD3+ decline and disease progression (33) suggests that the ability to detect X4 viruses early (or to predict their evolution) has clinical prognostic value. Since (as we have shown for eight individuals developing predicted X4 virus) PSSM scores tend to rise gradually, in contrast to the relatively abrupt appearance of X4 virus in the blood, as detected by biological phenotyping (28, 30), it is unlikely that X4 virus arises and outgrows R5 rapidly via single mutations. Monitoring the average score of subject virus (via sequencing or less costly methods) can provide advance warning of X4 outgrowth before virus is actually able to use CXCR4, which can in turn inform prognosis or treatment decisions. In support of this, a recently presented prospective study of 1,107 HIV-positive individuals starting suppressive antiretroviral therapy showed that the presence of SI viruses at the baseline (as predicted from consensus V3 sequences by PSSM and 11/25 methods) was an independent predictor of rapid CD4+ T-cell decline and mortality on therapy (Harrigan et al., 2nd Int. AIDS Soc. Conf. HIV Pathogenesis and Treatment). Evolutionary analysis of serially sampled viral sequences might allow us to identify the order in which mutations occur and highlight mutations that typically occur early in the R5-X4 transition. A preliminary analysis of the mutational pathways inferred for the individuals we studied suggests that basic mutations at sites 11 and 25 (as well as basic changes appearing at site 32) consistently occur early in the evolution of high-scoring lineages. The detection of these mutations in infected individuals may indicate a high X4 risk going forward, even when X4 virus is not yet present; larger-scale longitudinal sequencing studies are required to answer this question definitively.
While X4 virus generally develops once and relatively gradually in an individual, as previously suggested by van ’t Wout et al. (62), our phylogenetic analyses suggest that ultimately, it can be lost in two different ways: either by being supplanted by a preexisting population of R5 virus or by evolutionary reversion to the R5 phenotype. In the former case, R5 virus lineages from earlier in infection persist throughout infection, while in the latter, early R5 lineages are extinguished. This suggests that at least two qualitatively different types of R5 virus (or host responses to R5 virus) can occur in vivo. This idea parallels early observations (1, 13, 65) and a recent study (49) suggesting that NSI virus late-growth characteristics are different from those of the NSI virus that tends to initiate infection. A persistent R5 population may have evolved more efficient binding to the CCR5 receptor, as has been shown to occur with in vitro-passaged virus under pressure from a small-molecule CCR5 inhibitor (60), making it better able to exploit diminishing resources. Reverted X4 lineages, on the other hand, might retain the ability to use CXCR4 despite relatively low PSSM scores. The selective forces that lead to either of these or other outcomes will vary with host-specific factors but may also involve differences in the viral genetic background. By providing reconstructed amino acid sequences for the ancestral V3 sequences (i.e., sequences at the internal nodes of the tree) that can be expressed and used as reagents in in vitro experiments (4), phylogenetic analysis allows hypotheses such as these to be tested.
The PSSM score is a simple yet reliable method for predicting viral phenotypes on the basis of the amino acid sequence of the V3 loop of env. Such determinations are made on the basis of an additive model of CXCR4-usage propensity that ignores length variation and possible synergistic effects that certain residues at multiple sites in cis can have on phenotypes. Nevertheless, the method is robust with respect to these shortcomings (Table 1) and as a predictor of CXCR4 usage performs in a manner comparable to that of the neural network method (42) (sensitivity, 75%; specificity, 94%), which does incorporate synergistic effects. This suggests that amino acid residues at particular sites in V3 contribute (mostly independently) to coreceptor usage regardless of their particular combination in the haplotype. The PSSM method also has the advantage of being simpler in concept and more transparent in its assumptions than other methods (apart from the charged-based method) that have been previously employed (see Jensen and van ′t Wout [24] for a review of current methods). The PSSM score is a bioinformatic tool, complementing biological phenotype determination, that can express the X4 potential of a given V3 loop sequence in a graded way and for which intermediate values appear to correspond well with the evolution of viruses within individuals. As such, it may be useful as a basis of sequence-based clinical assays of within-host X4 outgrowth and could allow longitudinal study of X4 evolution and disease in large numbers of individuals without requiring extensive cloning of primary viral isolates.
Acknowledgments
This work benefited greatly from the comments of Geoff Gottlieb, Ron Swanstrom, and two anonymous reviewers.
This investigation was supported by grants from the university of Washington Center for AIDS Research. R.S. is supported by funds from Allegheny-Singer Research Institute. M.A.J. was supported by an NI-AID STD/AIDS training grant.
Appendix
Assume that in an epidemic, all new infections are caused by R5 viruses, and all X4 viruses result from within-individual HIV evolution. A simple model of the average yearly dynamics of X4 and R5 virus in an epidemic is then given by the following pair of equations:
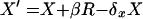 |
(5) |
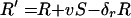 |
(6) |
where X is the number of X4-harboring individuals, R is the number of R5-harboring individuals, S is the number of susceptibles, all averaged per year, the primed variables are the corresponding number in the following year, v is the population rate of infection, β is the rate of conversion from R5 to X4 virus within an individual per year, and δx and δr are the rates of death in X4- and R5-harboring individuals, respectively.
Assume that an epidemic is at steady-state, such that the numbers of infected and susceptible individuals are unchanging over time at the prevailing levels of transmission. Then S = Ŝ is unchanging, and the steady-state number of X4-harboring individuals is found by setting X = X′ = X̂ and R = R′ = R̂ in the above equations and solving. We find that
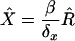 |
(7) |
and the steady-state fraction of X4-harboring individuals is
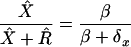 |
(8) |
In the text, since the severely ill are unlikely to be contributing to the infection of susceptible individuals, we estimated X4 prevalence using the AIDS rate, and not the death rate, of X4-harboring individuals. However, when we use an estimate of X4 death rate (δx) from Kupfer et al. (31) of 3.9%/yr, our estimate of X4 prevalence jumps to 54%. This is within the often-quoted range of 50%-60% of individuals who ultimately develop X4 virus, as assayed by phenotype testing, though the subjects investigated in Kupfer et al. had a median CD4+ T-cell count of 120/μl, well below the clinical cutoff value for AIDS.
Note that all per-year estimates of rates were extrapolated assuming a proportional decline per year. That is, when a death rate of R% over t years is reported, then we estimated the per-year rate δ as
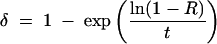 |
(9) |
rather than the simple R/t.
REFERENCES
- 1.Åsjö, B., L. Morfeldt-Manson, J. Albert, G. Biberfeld, A. Karlsson, K. Lidman, and E. M. Fenyö. 1986. Replicative capacity of human immunodeficiency virus from patients with varying severity of HIV infection. Lancet ii:660-662. [PubMed]
- 2.Björndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyö. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaak, H., A. B. van't Wout, M. Brouwer, B. Hooibrink, E. Hovenkamp, and H. Schuitemaker. 2000. In vivo HIV-1 infection of CD45RA+CD4+ T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4+ T cell decline. Proc. Natl. Acad. Sci. USA 97:1269-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, B. S., and M. J. Donoghue. 2000. Recreating ancestral proteins. Trends Ecol. Evol. 15:109-114. [DOI] [PubMed] [Google Scholar]
- 5.Chesebro, B., J. Nishio, S. Perryman, A. Cann, W. O'Brien, I. S. Y. Chen, and K. Wehrly. 1991. Identification of human immunodeficiency virus envelope gene sequences influencing viral entry into CD4-positive HeLa cells, T-leukemic cells, and macrophages. J. Virol. 65:5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chesebro, B., K. Wehrly, J. Nishio, and S. Perryman. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J. Virol. 66:6547-6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chesebro, B., K. Wehrly, J. Nishio, and S. Perryman. 1996. Mapping of independent V3 envelope determinants of human immunodeficiency virus type 1 macrophage tropism and syncytium formation in lymphocytes. J. Virol. 70:9055-9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connor, R. I., K. E. Sheridan, D. Ceradini, S. Choe, and N. R. Landau. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185:621-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Jong, J. J., A. De Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66:6777-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jong, J. J., J. Goudsmit, W. Keulen, B. Klaver, W. Krone, M. Tersmette, and A. de Ronde. 1992. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J. Virol. 66:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Roda Husman, A. M., R. P. van Rij, H. Blaak, S. Broersen, and H. Schuitemaker. 1999. Adaptation to promiscuous usage of chemokine receptors is not a prerequisite for human immunodeficiency virus type 1 disease progression. J. Infect. Dis. 180:1106-1115. [DOI] [PubMed] [Google Scholar]
- 12.de Wolf, F., J. Goudsmit, D. A. Paul, J. M. Lange, C. Hooijkaas, P. Schellekens, R. A. Coutinho, and J. van der Noordaa. 1987. Risk of AIDS related complex and AIDS in homosexual men with persistent HIV antigenaemia. Br. Med. J. 295:569-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenyö, E. M., J. Albert, and B. Åsjö. 1989. Replicative capacity, cytopathic effect and cell tropism of HIV. AIDS 3(Suppl. 1):S5-S12. [DOI] [PubMed] [Google Scholar]
- 14.Fenyö, E. M., L. Morfeldt-Månson, F. Chiodi, B. Lind, A. Von Gegerfelt, J. Albert, E. Olausson, and B. Åsjö. 1988. Distinct replicative and cytopathic characteristics of human immunodeficiency virus isolates. J. Virol. 62:4414-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fouchier, R. A., M. Brouwer, S. M. Broersen, and H. Schuitemaker. 1995. Simple determination of human immunodeficiency virus type 1 syncytium-inducing V3 genotype by PCR. J. Clin. Microbiol. 33:906-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fouchier, R. A. M., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gange, S. J., A. Muñoz, J. S. Chmiel, A. D. Donnenberg, L. M. Kirstein, R. Detels, and J. B. Margolick. 1998. Identification of inflections in T-cell counts among HIV-1-infected individuals and relationship with progression to clinical AIDS. Proc. Natl. Acad. Sci. USA 95:10848-10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glushakova, S., Y. Yi, J. C. Grivel, A. Singh, D. Schols, E. De Clercq, R. G. Collman, and L. Margolis. 1999. Preferential coreceptor utilization and cytopathicity by dual-tropic HIV-1 in human lymphoid tissue ex vivo. J. Clin. Investig. 104:R7-R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gribskov, M., A. D. McLachlan, and D. Eisenberg. 1987. Profile analysis: detection of distantly related proteins. Proc. Natl. Acad. Sci. USA 84:4355-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henikoff, S., J. C. Wallace, and J. P. Brown. 1990. Finding protein similarities with nucleotide sequence databases. Methods Enzymol. 183:111-132. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman, N. G., F. Seillier-Moiseiwitsch, J. Ahn, J. M. Walker, and R. Swanstrom. 2002. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J. Virol. 76:3852-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung, C. S., S. Pontow, and L. Ratner. 1999. Relationship between productive HIV-1 infection of macrophages and CCR5 utilization. Virology 264:278-288. [DOI] [PubMed] [Google Scholar]
- 23.Hwang, S. S., T. J. Boyle, H. Kim-Lyerly, and B. R. Cullen. 1991. Identification of the envelope v3 loop as the primary determinant of cell tropism in HIV-1. Science 253:71-74. [DOI] [PubMed] [Google Scholar]
- 24.Jensen, M. A., and A. van 't Wout. 2003. Predicting HIV-1 coreceptor usage using sequence analysis. AIDS Rev. 5:104-112. [PubMed] [Google Scholar]
- 25.Kaslow, R. A., D. G. Ostrow, R. Detels, J. P. Phair, B. F. Polk, and C. R. Rinaldo, Jr. 1987. The Multicenter AIDS Cohort Study: rationale, organization, and selected characteristics of the participants. Am. J. Epidemiol. 126:310-318. [DOI] [PubMed] [Google Scholar]
- 26.Koot, M., I. Keet, A. Vos, R. deGoede, M. Roos, R. Coutinho, F. Miedema, P. Schellekens, and M. Tersmette. 1993. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 118:681-688. [DOI] [PubMed] [Google Scholar]
- 27.Koot, M., R. van Leeuwen, R. E. de Goede, I. P. Keet, S. Danner, J. K. Eeftinck Schattenkerk, P. Reiss, M. Tersmette, J. M. Lange, and H. Schuitemaker. 1999. Conversion rate towards a syncytium-inducing (SI) phenotype during different stages of human immunodeficiency virus type 1 infection and prognostic value of SI phenotype for survival after AIDS diagnosis. J Infect. Dis. 179:254-258. [DOI] [PubMed] [Google Scholar]
- 28.Koot, M., A. H. Vos, R. P. Keet, R. E. de Goede, M. W. Dercksen, F. G. Terpstra, R. A. Coutinho, F. Miedema, and M. Tersmette. 1992. HIV-1 biological phenotype in long-term infected individuals evaluated with an MT-2 cocultivation assay. AIDS 6:49-54. [DOI] [PubMed] [Google Scholar]
- 29.Kozak, S. L., E. J. Platt, N. Madani, J. Ferro, F. E., K. Peden, and D. Kabat. 1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol. 71:873-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuiken, C. L., J. J. de Jong, E. Baan, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Evolution of the V3 envelope domain in proviral sequences and isolates of human immunodeficiency virus type 1 during transition of the viral biological phenotype. J. Virol. 66:5704. [PMC free article] [PubMed] [Google Scholar]
- 31.Kupfer, B., R. Kaiser, J. K. Rockstroh, B. Matz, and K. E. Schneweis. 1998. Role of HIV-1 phenotype in viral pathogenesis and its relation to viral load and CD4+ T-cell count. J. Med. Virol. 56:259-263. [PubMed] [Google Scholar]
- 32.Lupas, A. 1996. Coiled coils: new structures and new functions. Trends Biochem. Sci. 21:375-382. [PubMed] [Google Scholar]
- 33.Maas, J. J., S. J. Gange, H. Schuitemaker, R. Coutinho, R. van Leeuwen, and J. B. Margolick. 2000. Strong association between failure of T cell homeostasis and the syncytium-inducing phenotype among HIV-1-infected men in the Amsterdam Cohort Study. AIDS 14:1155-1161. [DOI] [PubMed] [Google Scholar]
- 34.Manly, B. F. J. 1997. Randomization, bootstrap and Monte Carlo methods in biology, 2nd ed. Chapman & Hall, London, United Kingdom.
- 35.McDonald, R. A., G. Chang, and N. L. Michael. 2001. Relationship between V3 genotype, biologic phenotype, tropism, and coreceptor use for primary isolates of human immunodeficiency virus type 1. J. Hum. Virol. 4:179-187. [PubMed] [Google Scholar]
- 36.Miedema, F., L. Meyaard, M. Koot, M. R. Klein, M. T. L. Roos, M. Groenink, R. A. M. Fouchier, A. B. van ’t Wout, M. Tersmette, P. T. A. Schellekens, and H. Schuitemaker. 1994. Changing virus-host interactions in the course of HIV-1 infection. Immunol. Rev. 140:35-72. [DOI] [PubMed] [Google Scholar]
- 37.Milich, L., B. Margolin, and R. Swanstrom. 1993. V3 loop of the human immunodeficiency virus type 1 Env protein: interpreting sequence variability. J. Virol. 67:5623-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson, J. A., F. Baribaud, T. Edwards, and R. Swanstrom. 2000. Patterns of changes in human immunodeficiency virus type 1 V3 sequence populations late in infection. J. Virol. 74:8494-8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Philpott, S., B. Weiser, K. Anastos, C. M. Kitchen, E. Robison, W. A. Meyer III, H. S. Sacks, U. Mathur-Wagh, C. Brunner, and H. Burger. 2001. Preferential suppression of CXCR4-specific strains of HIV-1 by antiviral therapy. J. Clin. Investig. 107:431-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pillai, S., B. Good, D. Richman, and J. Corbeil. 2003. A new perspective on V3 phenotype prediction. AIDS Res. Hum. Retrovir. 19:145-149. [DOI] [PubMed] [Google Scholar]
- 41.Reche, P. A., J. P. Glutting, and E. L. Reinherz. 2002. Prediction of MHC class I binding peptides using profile motifs. Hum. Immunol. 63:701-709. [DOI] [PubMed] [Google Scholar]
- 42.Resch, W., N. Hoffman, and R. Swanstrom. 2001. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 288:51-62. [DOI] [PubMed] [Google Scholar]
- 43.Richman, D. D., and S. A. Bozzette. 1994. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J Infect. Dis. 169:968-974. [DOI] [PubMed] [Google Scholar]
- 44.Rinaldo, C. R., Jr., P. Gupta, X. L. Huang, Z. Fan, J. I. Mullins, S. Gange, H. Farzadegan, R. Shankarappa, A. Munoz, and J. B. Margolick. 1998. Anti-HIV type 1 memory cytotoxic T lymphocyte responses associated with changes in CD4+ T cell numbers in progression of HIV type 1 infection. AIDS Res. Hum. Retrovir. 14:1423-1433. [DOI] [PubMed] [Google Scholar]
- 45.Ross, H. A., and A. G. Rodrigo. 2002. Immune-mediated positive selection drives human immunodeficiency virus type 1 molecular variation and predicts disease duration. J. Virol. 76:11715-11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rübsamen-Waigmann, H., W. B. Becker, E. B. Helm, R. Brodt, H. Fischer, K. Henco, and H. D. Brede. 1986. Isolation of variants of lymphocytopathic retroviruses from the peripheral blood and cerebral spinal fluid of patients with ARC or AIDS. J. Med. Virol. 19:335-344. [DOI] [PubMed] [Google Scholar]
- 47.Scarlatti, G., E. Tresoldi, A. Björndal, R. Fredriksson, C. Colognesi, H. K. Deng, M. S. Malnati, A. Plebani, A. G. Siccardi, D. R. Littman, E. M. Fenyö, and P. Lusso. 1997. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 3:1259-1265. [DOI] [PubMed] [Google Scholar]
- 48.Schuitemaker, H., N. Kootstra, R. de Goede, F. de Wolf, F. Miedema, and M. Tersmette. 1991. Monocytotropic human immunodeficiency virus type 1 (HIV-1) variants detectable in all stages of HIV-1 infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J. Virol. 65:356-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scoggins, R. M., J. R. Taylor, Jr., J. Patrie, A. B. van ’t Wout, H. Schuitemaker, and D. Camerini. 2000. Pathogenesis of primary R5 human immunodeficiency virus type 1 clones in SCID-hu mice. J. Virol. 74:3205-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X.-L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shioda, T., J. A. Levy, and C. Cheng-Mayer. 1991. Macrophage and T cell-line tropisms of HIV-1 are determined by specific regions of the envelope gp120 gene. Nature 349:167-169. [DOI] [PubMed] [Google Scholar]
- 52.Simmons, G., D. Wilkinson, J. D. Reeves, M. T. Dittmar, S. Beddows, J. Weber, G. Carnegie, U. Desselberger, P. W. Gray, R. A. Weiss, and P. R. Clapham. 1996. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J. Virol. 70:8355-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh, A., and R. G. Collman. 2000. Heterogeneous spectrum of coreceptor usage among variants within a dualtropic human immunodeficiency virus type 1 primary-isolate quasispecies. J. Virol. 74:10229-10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71:7136-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stormo, G. D. 1988. Computer methods for analyzing sequence recognition of nucleic acids. Annu. Rev. Biophys. Biophys. Chem. 17:241-263. [DOI] [PubMed] [Google Scholar]
- 56.Swofford, D. L. 1999. PAUP* 4.0: phylogenetic analysis using parsimony (and other methods), version 4.0b2a. Sinauer Associates, Inc., Sunderland, Mass.
- 57.Tersmette, M., R. E. Y. de Goede, B. J. M. Al, I. N. Winkel, R. A. Gruters, H. T. Cuypers, H. G. Huisman, and F. Miedema. 1988. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J. Virol. 62:2026-2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tersmette, M., R. A. Gruters, F. de Wolf, R. E. de Goede, J. M. Lange, P. T. Schellekens, J. Goudsmit, H. G. Huisman, and F. Miedema. 1989. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J. Virol. 63:2118-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trkola, A., S. E. Kuhmann, J. M. Strizki, E. Maxwell, T. Ketas, T. Morgan, P. Pugach, S. Xu, L. Wojcik, J. Tagat, A. Palani, S. Shapiro, J. W. Clader, S. McCombie, G. R. Reyes, B. M. Baroudy, and J. P. Moore. 2002. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. USA 99:395-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Rij, R. P., H. Blaak, J. A. Visser, M. Brouwer, R. Rientsma, S. Broersen, A. M. de Roda Husman, and H. Schuitemaker. 2000. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J. Clin. Investig. 106:1039-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van 't Wout, A. B., H. Blaak, L. J. Ran, M. Brouwer, C. Kuiken, and H. Schuitemaker. 1998. Evolution of syncytium-inducing and non-syncytium-inducing biological virus clones in relation to replication kinetics during the course of human immunodeficiency virus type 1 infection. J. Virol. 72:5099-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van 't Wout, A. B., N. A. Kootstra, G. A. Mulder-Kampinga, N. Albrecht-van Lent, H. J. Scherpbier, J. Veenstra, K. Boer, R. A. Coutinho, F. Miedema, and H. Schuitemaker. 1994. Macrophage-tropic variants initiate HIV-1 infection after sexual, parenteral, and vertical transmission. J. Clin. Investig. 94:2060-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vodicka, M. A., W. C. Goh, L. I. Wu, M. E. Rogel, S. R. Bartz, V. L. Schweickart, C. J. Raport, and M. Emerman. 1997. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 233:193-198. [DOI] [PubMed] [Google Scholar]
- 65.von Briesen, H., W. B. Becker, K. Henco, E. B. Helm, H. R. Gelderblom, H. D. Brede, and H. Rübsamen-Waigmann. 1987. Isolation frequency and growth properties of HIV-variants: multiple simultaneous variants in a patient demonstrated by molecular cloning. J. Med. Virol. 23:51-66. [DOI] [PubMed] [Google Scholar]
- 66.Xiao, L., S. M. Owen, I. Goldman, A. A. Lal, J. J. deJong, J. Goudsmit, and R. B. Lal. 1998. CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 is independent of phylogenetically distinct global HIV-1 isolates: delineation of consensus motif in the V3 domain that predicts CCR-5 usage. Virology 240:83-92. [DOI] [PubMed] [Google Scholar]
- 67.Zhang, L. Q., P. MacKenzie, A. Cleland, E. C. Holmes, A. J. Leigh Brown, and P. Simmonds. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol. 67:3345-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu, T., H. Mo, N. Wang, D. S. Nam, Y. Cao, R. A. Koup, and D. D. Ho. 1993. Genotypic and phenotypic characterization of HIV-1 in patients with primary infection. Science 261:1179-1181. [DOI] [PubMed] [Google Scholar]