


Abstract
In eukaryotes with the universal genetic code a single class I release factor (eRF1) most probably recognizes all stop codons (UAA, UAG and UGA) and is essential for termination of nascent peptide synthesis. It is well established that stop codons have been reassigned to amino acid codons at least three times among ciliates. The codon specificities of ciliate eRF1s must have been modified to accommodate the variant codes. In this study we have amplified, cloned and sequenced eRF1 genes of two hypotrichous ciliates, Oxytricha trifallax (UAA and UAG for Gln) and Euplotes aediculatus (UGA for Cys). We also sequenced/identified three protist and two archaeal class I RF genes to enlarge the database of eRF1/aRF1s with the universal code. Extensive comparisons between universal code eRF1s and those of Oxytricha, Euplotes and Tetrahymena, which represent three lineages that acquired variant codes independently, provide important clues to identify stop codon-binding regions in eRF1. Domain 1 in the five ciliate eRF1s, particulary the TASNIKS heptapeptide and its adjacent region, differs significantly from domain 1 in universal code eRF1s. This observation suggests that domain 1 contains the codon recognition site, but that the mechanism of eRF1 codon recognition may be more complex than proposed by Nakamura et al. or Knight and Landweber.
INTRODUCTION
Variant genetic codes have arisen several times in prokaryotic and eukaryotic evolution (1). Where sense codons have been reassigned to different amino acids, modifications in codon recognition properties of tRNAs are characteristically involved (1). Where stop codons have been reassigned as sense codons, changes in the translation termination machinery, ensuring that these codons are no longer read as signals for peptide chain termination, are presumably also required. Although the possibility that rRNA might be involved in stop codon recognition is not excluded as yet, class I release factors (RFs), proteins which are thought to recognize stop codons directly, are the most likely site for such changes.
In bacteria, two class I RFs are involved in stop codon recognition: RF1 (which reads UAA and UAG) and RF2 (UAA and UGA) (2,3). Mycoplasma spp. that use the UGA codon as Trp no longer recognize this triplet as a termination signal (4), simply because they have lost RF2 (5,6). However, since eukaryotes and archaea use a single class I RF (eRF1 and aRF1, respectively) to read all three stop codons (2,3,7,8), the evolution of alternative codes in these organisms has presumably entailed changes in the structure of the protein.
Although the sequence similarity between bacterial (RF1 and RF2) and eukaryotic class I RFs is difficult to detect, both are thought to be ‘molecular mimics’, resembling tRNAs in function and structure (3,9). Indeed, mutational studies have identified specific tripeptide ‘anticodons’ in bacterial RF1 and RF2 (10). Such studies are less well advanced in eukaryotes and a comparison of the sequences of eRF1s from eukaryotes with the universal and variant codes might provide the first clues as to how codon recognition functions map to elements of protein structure. Fifteen (complete) eRF1 sequences are available in the SWISSPROT database as of September 2000: all are from organisms using the universal code except that from Tetrahymena thermophila (which reads UAR as Gln, where R = A or G) (11).
Structural studies of human eRF1 suggest that while the middle domain (domain 2) of eRF1 is involved in peptidyl-tRNA hydrolysis, the N-terminal domain (domain 1) of this protein may be analogous to the anticodon stem–loop of tRNA (12). Nakamura et al. (13) have recently nominated a Thr-Ala-Ser (TAS) tripeptide within this domain as a potential ‘omnipotent discriminator’, because it was found in all eukaryotic sequences available to them except that of T.thermophila [which has Lys-Ala-Ser (KAS) at this position]. Alternatively, Knight and Landweber suggested that a tetrapeptide immediately C-terminal to TAS [Asn-Ile-Lys-Ser (NIKS) in all eukaryotes except for T.thermophila, which has Asn-Ile-Lys-Asp] is crucial in stop codon recognition (14). Unlike the TAS triplet, Asn-Ile-Lys (NIK) is also highly conserved in aRF1s and recent in vitro studies indicated that aRF1 and eRF1 recognize stop codons in a very similar way (8).
Additional eRF1 and aRF1 sequences, especially eRF1s from other eukaryotes with variant codes acquired independently, should allow refinement of inferences about the involvement of specific residues in stop codon recognition. In this study we have cloned and sequenced the eRF1 genes of ciliates with variant genetic codes, Oxytricha trifallax and Euplotes aediculatus (UAR for Gln and UGA for Cys, respectively). While the universal code is likely ancestral for ciliates, the distribution of alternative codes across the ciliate phylogeny suggests that such codes have arisen several times (15). Oxytricha and Euplotes, both hypotrichous ciliates, have different codes. Tetrahymena (UAR for Gln) belongs to the genus oligohymenophorea, not closely related to either Oxytricha and Euplotes (15). We also determined the entire coding region of the T.thermophila eRF1, previously known only as a cDNA sequence (11). In addition, we further expanded the database of eRF1s and aRF1s from species with the universal code by sequencing a Dictyostelium discoideum eRF1 cDNA and including DNA sequences newly identified among public databases. Our extensive analyses, including the five eRF1s of ciliates, which represent three independent code conversions, suggest the involvement of domain 1 in stop codon recognition.
MATERIALS AND METHODS
Amplification, cloning and sequencing of protist eRF1 genes
Macronuclear DNA samples of O.trifallax and E.aediculatus were provided by D.M. Prescott (University of Colorado, Boulder, CO). A set of degenerate PCR primers, 45F2A (CCTAAAAAGCATGGNCGNGGNGG, where N = A, C, G or T) and 45R3A (TTAATGCCGTTYTCNCCNCCRTA, Y = T or C), was utilized to amplify a portion of the ciliate eRF1 genes (0.3 kb). PCR was conducted as follows: denaturation at 94°C for 30 s, annealing at 40°C for 1 min and extension at 72°C for 1 min (35 cycles). Amplified fragments were cloned into vector pCR 2.1 TOPO (Invitrogen, Carlsbad, CA) and sequenced on both strands. Macronuclei in hypotrichous ciliates, including Oxytricha and Euplotes, are known to contain minichromosomes, which comprise a single gene and telomere ends with a repeat sequence of CCCCAAAA (16). We succeeded in amplifying an entire chromosome containing an eRF1 gene in two pieces with exact match PCR primers to the 0.3 kb amplified fragment and primers designed to anneal to telomere ends. Thermal cycling (35 cycles) was conducted by denaturation at 94°C for 1 min, annealing at 40°C for 30 s and extension at 72°C for 30 s. To avoid PCR artifacts, more than three clones were sequenced for each amplified fragment as described above. Primers for telomere ends of hypotrich macronuclear chromosomes were provided by J.M.Logsdon (Emory University, Atlanta, GA).
To assess the intron distribution in the T.thermophila eRF1 gene, a set of primers, Tt45F4 (AAATAGTTAGCGTAAATGGAAGAG) and Tt45R3 (CGAAGAAGAAGAAGGCTTCATATG), were prepared by reference to its cDNA sequence (AB026195). Using this set of primers the entire protein coding region of the Tetrahymena gene was amplified, cloned and completely sequenced on both strands.
The D.discoideum eRF1 cDNA clone SLI621 was kindly provided by the Dictyostelium cDNA Project (Tsukuba University, Tsukuba, Japan) (17) and was subsequently sequenced on both strands.
The amino acid sequence of the eRF1 encoded in the (eukaryotic) nuclear genome of a secondary endosymbiont (nucleomorph) of the cryptomonad Guillardia theta was generously provided by S.Douglas (Institute for Marine Bioscience, Halifax, Canada).
The aRF1 amino acid sequence of Methanosarcina mazei was provided by the Göttingen Genomic Laboratory of the University of Göttingen (Göttingen, Germany). The aRF1 amino acid sequence of Halobacterium sp. NRC-1 was provided by S.DasSarma (University of Massachusetts, Amherst, MA) prior to publication.
Identification of novel eRF1 and aRF1 genes in public databases
We surveyed unidentified protist eRF1 and aRF1 genes in public databases. In the GenBank non-redundant database at the National Institute for Biotechnology and Information we found the Oxytricha nova macronuclear sequence AF188150, which shows high similarity to eRF1s, using TBLASTN (18) and the amino acid sequence of the Giardia lamblia eRF1 (AF198107) as the query. The aRF1 homolog of Pyrococcus furiosus was retrieved from the World Wide Web site of the Center of Marine Biotechnology at University of Maryland (http://combdna.umbi.umd.edu/bags.html).
Phylogenetic analysis of eRF1 and aRF1
Newly determined or identified eRF1s and aRF1s were added manually to our previous alignment. From this master alignment composed of the 15 universal code eRF1s, five ciliate eRF1s and 10 aRF1s (the 15-5-10 dataset), all ambiguously aligned sites and sites containing more than one gap were removed and a total of 345 sites were used for phylogenetic analysis. An eRF1–aRF1 phylogeny was reconstructed under protein maximum likelihood criteria with the JTT amino acid substitution model incorporating among-site rate variation (one invariable category and the discrete γ distribution approximated with eight variable categories) implemented in PUZZLE v.4.0.2 (19). PUZZLE also provided a distance matrix incorporating among-site rate variation and so we reconstructed a distance tree based on this matrix using FITCH in PHYLIP v.3.572 (20).
Tree topologies were statistically evaluated as percent occurrence in 10 000 quartet puzzling trees. Further, eRF1–aRF1 trees were examined by resampling datasets: 500 replicates were generated from the 15-5-10 dataset using SEQBOOT in PHYLIP (20) and then distance matrices considering among-site rate variation were calculated from each replicate using PUZZLEBOOT v.1.02 (A.J.Roger and M.E.Holder, http://members.tripod.de/korbi/puzzle/) and PUZZLE (19). The proportion of invariable sites and the γ distribution α shape parameter were estimated to be 0.04 and 1.83, respectively. Bootstrap support values were obtained from 500 matrices using FITCH and CONSENSE in PHYLIP (20).
Calculation of relative amino acid substitution rates in eRF1
To calculate the relative amino acid substitution rate of each site in eRF1, three sub-datasets were generated from the 15-5-10 dataset. The 15-5-0 or 15-0-10 datasets were generated from the 15-5-10 dataset by excluding the 10 aRF1s or the five ciliate eRF1s, respectively. Subsequently, the five ciliate sequences were removed from the 15-5-0 dataset to create the 15-0-0 dataset. After polishing these alignments, 400 sites remained for the 15-0-0 and 15-5-0 datasets, while the 15-5-10 and 15-0-10 datasets had 384 sites. For each edited alignment a discrete γ distribution approximated with eight site rate categories (plus one invariable category) was separately calculated over a neighbor joining tree with the JTT model using PUZZLE (19). The averages of relative rates of a sliding window with 10 sites were calculated over the entire eRF1. The substitution rates of sites excluded from the analyses (gaps or ambiguously aligned sites) were arbitrarily adjudged as the fastest at 2.9989 (category 8).
RESULTS AND DISCUSSION
Characteristics of novel eRF1 and aRF1 genes
The macronuclear chromosome that contains the O.trifallax eRF1 gene was 1619 bp in length (not including telomere ends). The putative protein should have 445 residues, and four TAA and two TAG codons were observed in the presumed coding region. A short intron (39 bp) was found in the N-terminus (Fig. 2A). The best BLASTX match to the O.trifallax gene was the CG5605 gene product (eRF1) of Drosophila melanogaster (AE003591), with an expected (E) value of 10–152.
Figure 2.
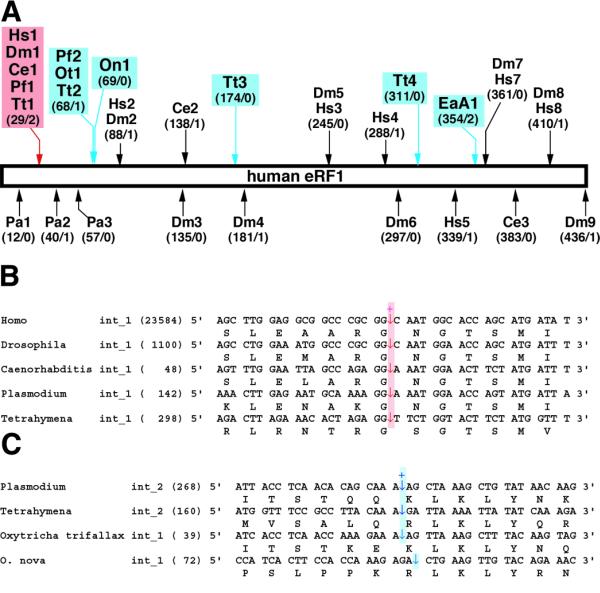
Introns in eRF1 genes. (A) Intron distribution in eRF1 genes. Intron positions are schematically mapped on the human eRF1 gene. The number of the corresponding residue where an intron is inserted (human eRF1 numbering on left) and the phase of the intron (0, 1 or 2 on right) are shown in parentheses. Hs, H.sapiens; Ce, C.elegans; Dm, D.melanogaster; Pa, Podospora anserina; Pf, Plasmodium falciparum; Ot, O.trifallax; On, O.nova; EaA, E.aediculatus A; Tt, T.thermophila. (B) Alignment of the region containing intron 1 of the human eRF1 gene. The intron position is indicated by arrows. The length of each intron is given in parentheses. (C) Alignment of the region containing intron 2 of the Plasmodium eRF1 gene. Details as described above.
From E.aediculatus we isolated two eRF1 genes in this study. The macronuclear chromosomes for the Euplotes A and B genes were 1508 and 1436 bp (not including telomere ends), respectively. Both putative genes encode 437 amino acid proteins, and two and four TGA codons were found in the coding regions of the A and B genes, respectively. Only the A gene appears to contain an intron (39 bp), located near its C-teminus (Fig. 2A). The best BLASTX matches to the Euplotes A and B genes in the GenBank non-redundant database were with the Arabidopsis thaliana eRF1 (P35614) and D.melanogaster eRF1 (AE003591), respectively (E values were both 10–140).
The protein coding region of the T.thermophila eRF1 (2468 bp) was predicted to have four introns, 298, 160, 463 and 243 bp in length. The DNA sequence amplified and sequenced in this study was identical to its cDNA sequence (AB026195) except for four intron sequences.
The cDNA for the D.discoideum eRF1 appeared to be 1657 bp in length and to cover the entire coding region of 441 residues (1323 bp). Comparing the cDNA sequence to genomic sequences retrieved from the D.discoideum Genome Project at the Institute of Molecular Biotechnology at Jena (http://genome.imb-jena.de/dictyostelium) indicates that this gene harbors no intron (data not shown). The highest BLASTX match to this sequence was the Xenopus laevis eRF1 (P35615), with an E value of 10–176.
We also found two previously unannotated eRF1/aRF1 genes in public databases. A macronucleus chromosome DNA sequence (AF188150) from O.nova shared strong similarity to eRF1s. The best BLASTX match against the GenBank non-redundant database was with the D.melanogaster eRF1 (AE003591), with an E value of 10–117. Our assessment revealed that this chromosome covers the entire eRF1 gene of 1374 bp (434 amino acids), with a 72 bp intron in the N-terminal region (Fig. 2A). A putative P.furiosus protein with 421 residues (ORF ID Pf_1487344, positions 1487344–1488606) matched the Pyrococcus abysii aRF1 (E75177), with an E value of 0.0, during a BLASTP search against the GenBank non-redundant database.
eRF1–aRF1 phylogeny and intron distribution in eRF1 genes
Our protein phylogenies inferred from the 15-5-10 dataset (15 universal code eRF1s, five ciliate eRF1s and 10 aRF1s) generally agree with previous analyses with limited sequence sampling (21): eRF1s and aRF1s obviously belong to the same protein family, but the branch length connecting the eukaryotic and archaeal clades is extremely long (Fig. 1). The five ciliate eRF1s form a monophyletic cluster [bootstrap/quartet puzzling (QP) scores 73/69; Fig. 1]. Within this clade the monophyly of both the Euplotes and Oxytricha sequences were statistically supported, but our analysis did not resolve deeper phylogenetic branching within the ciliates; the Oxytricha and Euplotes clades were weakly connected only in a distance analysis (bootstrap score 61; Fig. 1). The Dictyostelium eRF1 showed no strong affinity to other sequences examined in this analysis. The Guillardia nucleomorph eRF1 appeared as the deepest branch among the eukaryotic sequences tested so far (bootstrap/QP scores 67/80; Fig. 1), but this sequence, as well as the Pyrobaculum and Archaeoglobus aRF1s, failed in a χ2 test that compared the amino acid composition to the frequency distribution assumed in the ML model in PUZZLE (19). We suspect that the position of the Guillardia nucleomorph sequence is an artifact of ‘long branch’ attraction and/or aberrant amino acid composition.
Figure 1.
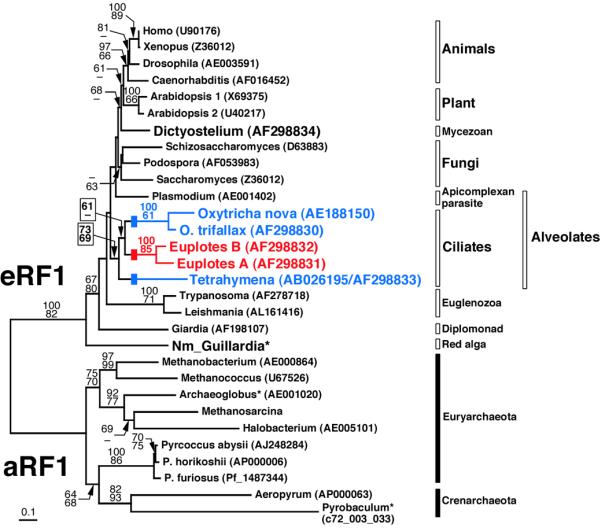
Phylogenetic tree inferred from eRF1 and aRF1 sequences. An alignment including the 15 universal code eRF1s, five variant code ciliate eRF1s and 10 aRF1s (the 15-5-10 dataset, 345 sites) was used. The tree was reconstructed from a distance matrix incorporating one invariable category and the discrete γ distribution approximated with eight variable categories, using FITCH in PHYLIP (20). Robustness of tree topologies was examined by bootstrap analysis (500 resamplings) using a distance method considering among-site substitution rates (top) and percent occurrence in 10 000 quartet puzzling trees (bottom). A dash indicates that a given node was not supported by the bootstrap score or quartet puzzling score (>60). An asterisk indicates sequences that failed in a χ2 test of amino acid composition in PUZZLE (19). The eRF1s from ciliates with variant codes are highlighted in blue (UAR for Gln) and red (UGA for Cys). Blocks on the Oxytricha, Euplotes and Tetrahymena branches indicate independent conversions from the universal code to variant codes. For the P.furiosus and Pyrobaculum aerophilum aRF1s their ORF IDs are listed, instead of GenBank accession numbers. Nm_Guillardia, Guillardia nucleomorph eRF1. The amino acid sequences of the Guillardia nucleomorph eRF1 and Methanosarcina aRF1 are available on request.
Introns were most abundant in the Tetrahymena eRF1 gene among the five ciliate genes tested so far (Fig. 2A). Tetrahymena intron 1 appears to be inserted in a position homologous to an intron position in animal (Homo sapiens, D.melanogaster and Caenorhabditis elegans) and Plasmodium falciparum (an apicomplexan parasite) eRF1 genes (Fig. 2A and B). These organisms are described as ‘crown eukaryotes’, mainly based on small subunit (SSU) rRNA phylogenies (22). Therefore this intron may predate the divergence of crown eukaryotes.
Ultrastructural observations and molecular phylogenies (mostly inferred from SSU rRNA sequences) unify ciliates, apicomplexan parasites (e.g. Plasmodium) and dinoflagellates into a single clade, Alveolata (23). In agreement with the taxonomy, three out of six alveolate eRF1 genes shared an intron position (Tetrahymena intron 2, Plasmodium intron 2 and O.trifallax intron 1; Fig. 2A and C). Thus the eRF1 gene of a common ancestor of ciliates and apicomplexan parasites may have harbored this intron. A further survey of introns in protist eRF1 genes, particulary dinoflagellate genes, would allow us to examine whether this intron is Alveolate-specific.
Although the two Oxytricha sequences were robustly tied in our trees (Fig. 1), the O.nova intron is not inserted in the same position as the O.trifallax intron; the positions are 2 bp apart from each other (Fig. 2A and C). No sequence similarity among the four intron sequences was detected (data not shown). However, we do not know whether this represents an instance of ‘intron sliding’ (24,25). No intron inserted in positions homologous to intron 3 and 4 in the Tetrahymena gene and intron 1 in the Euplotes A gene have been found in other genes studied (Fig. 2A).
Comparison of eRF1 sequences using relative substitution rate calculation
We calculated relative substitution rates using the dataset including 15 non-ciliate eRF1s (the 15-0-0 datase). Our analysis indicated that domain 3 is the most rapidly evolving overall within eukaryotes (Fig. 3A). The addition of 10 archaeal RF1s did not significantly alter substitution rates, but domain 3 is the most poorly conserved between eRF1s and aRF1s (data not shown). In particular, the two aRF1s of Pyrobaculum and Aeropyrum (Crenarchaeota) lack part of a highly diverged region in domain 3 (Fig. 3A). The rapid evolution of domain 3 observed in these analyses implies that the crucial determinant of eRF3 binding might be the secondary or tertiary structure of this domain.
Figure 3.

Comparison of eRF1s using relative amino acid substitution rates. (A) Distribution of relative substitution rates calculated from the datasets including the 15 universal code eRF1s (the 15-0-0 dataset, shown in black) or the universal code and five ciliate (variant code) eRF1s (the 15-5-0 dataset, shown in red). The relative substitution rates of individual sites were calculated by PUZZLE v.4.0.2 (19) under the JTT model considering one invariable category and the discrete γ distribution approximated with eight variable categories. Averages of relative rates of a sliding window with 10 sites were plotted over the entire eRF1. Although sites that are ambiguously aligned and include more than one gap were excluded from the site rate analyses, their rates were arbitrarily considered as the fastest at 2.9989 (category 8) for calculation of the 10 sites averages. The regions corresponding to α-helices and β-sheets identified in human eRF1 structure are indicated as closed and open boxes, respectively. Sites 1 and 421 correspond to Glu13 and Tyr443 in human eRF1, respectively. The five arrows on the x-axis indicate the residues corresponding to Ile32, Met48, Val68, Leu123 and His129 in yeast eRF1 (32). (B) Alignment of domain 1 (sites 31–91). Eight categories of relative substitution rates inferred from the 15-0-0 and 15-0-10 (15 universal code eRF1s and 10 aRF1s) datasets are listed in the top two lines. Sites estimated as invariable (category 0) from the analyses using either the 15-0-0 or 15-0-10 dataset are boxed. Sites excluded from rate analyses are indicated by ‘g’. A dot indicates identity with the corresponding position in human eRF1 (top row). Unique residues in the ciliate eRF1s which have not been found in the universal code eRF1s so far are highlighted in red. Guillardia (Nm), Guillardia nucleomorph eRF1. The regions corresponding to α-helices and the β2-sheet in human eRF1 structure are shown by closed and open boxes. (C) Alignment of domain 2 (sites 162–200). Details are as described above. (D) Comparison of the residues nominated as stop codon binding sites by Bertram et al. (32). The residues that correspond to Ile32, Met48, Val68, Leu123 and His129 (yeast eRF1 numbering) are aligned from left to right. A dot indicates identity with the corresponding position in yeast eRF1 (top row). Details as described above.
It has been shown that this domain is responsible for binding to eRF3, a GTPase which facilitates translation termination in eukaryotes (26–29). In particular, the acidic amino acid tails of budding and fission yeasts have been shown to be essential for the eRF1–eRF3 interaction (26–28). However, this region is not well conserved in other eukaryotes, with Leishmania and O.nova lacking any extensive acidic amino acid stretch (Fig. 4). The Guillardia nucleomorph eRF1 is the most severely truncated (Fig. 4). Nucleomorphs of cryptomonads are most probably descendants of a red alga (30) and the eRF1 of Porphyra yezoensis (a red alga) has a regular acidic amino acid tail (Fig. 4). We suggest that this ancestral nucleomorph eRF1 had a tail that was subsequently lost during genome reduction in the endosymbiont. It is also probable that the Leishmania and O.nova eRF1s lost the standard tails independently (Fig. 4).
Figure 4.

Comparison of the C-terminal region of eRF1s and aRF1s. Sites estimated as invariable (category 0) from the analyses using either the 15-0-0 or 15-0-10 dataset are boxed. Sites excluded from rate analyses are indicated by ‘g’. A dot indicates identity with the corresponding position in human eRF1 (top row). Sequences of the C-terminal tail of eRF1 are not aligned. Acidic residues in the C-terminal tail of eRF1, such as Asp (D), Glu (E), Asn (N) and Gln (Q), are highlighted in blue. The GenBank accession no. of the Porphyra yezoensis eRF1 cDNA is AV437473. NM_Guillardia, Guillardia nucleomorph eRF1.
In domain 2 it appears that a loop region between the β7-sheet and α5-helix is highly conserved (Fig. 3A) and this loop includes the GGQ motif considered essential for eRF1 function (12). The relative site rates of this domain were not affected by addition of the ciliate (variant code) eRF1s (Fig. 3A) and it is unlikely that the GGQ motif (and domain 2) contains a stop codon-binding site. In fact, biochemical studies using human eRF1 indicate that mutations of Gly residues in this motif specifically abolish peptidyl-tRNA hydrolysis (but not ribosome binding or induction of eRF3 GTPase activity) (31). Genetic studies with yeast confirm the conclusion that the GGQ motif promotes peptidyl-tRNA hydrolysis at the peptidyltransferase centre of the ribosome (12). This motif was completely conserved in all aRF1s and eRF1s, including those from ciliates with variant codes (Fig. 3C).
Domain 1 contains the TAS tripeptide and the adjacent tetrapeptide NIKS (NIK is also conserved in archaeal RF1s) (Fig. 3B). These two regions at the tip of domain 1 have been previously proposed as discriminators in stop codon recognition by eRF1, in part because they are conserved in all eRF1s except that of Tetrahymena, the single eRF1 characterized from an organism with a variant code before this work (13,14). Our substitution rate analyses indicate that, in this domain of universal code eRF1s (and aRF1s as well), regions around the TASNIKS heptapeptide are the most slowly evolving (Fig. 3A). However, substitution rates around this heptapeptide were significantly increased when the five ciliate eRF1s were incorporated into the rate analyses (Fig. 3A). In fact, all variant code eRF1s have substitutions in this heptapeptide stretch, which, in contrast, is completely conserved in universal code eRF1s, even the most divergent, that of the nucleomorph in Guillardia (Fig. 3B). This speaks strongly for the involvement of this heptapeptide in codon recognition. However, each of the variant code eRF1s has a different sequence in this region (Fig. 3B) and so the TASNIKS heptepeptide might be highly constrained to maintain the tertiary structure of universal code eRF1s (particulary domain 1). Structural constraints affecting the heptapeptide in the ciliate eRF1s might be relaxed to accommodate their variant codes. So far it seems unlikely that a simple codon–anticodon type discrimination, as proposed for bacterial RF1 and RF2 (10), will be obtained for eRF1 and aRF1.
A recent genetic study using yeast eRF1 and molecular modeling of human eRF1 and the UAA triplet indicated that residues outside the TASNIKS heptapeptide, Ile32, Met48, Val68, Leu123 and His129 (yeast eRF1 numbering), were involved in stop codon binding (32). Interestingly, these residues are scattered in the primary structure of domain 1 (Fig. 3A), but they are neighbors in the tertiary structure (32). The corresponding residues in the five ciliate proteins, particulary Leu123 and His129 in yeast eRF1, appear to diverge (Fig. 3D). However, further biochemical, genetic and structural studies are required to test hypotheses of stop codon binding by eRF1. In addition, eRF1s from additional lineages that acquired a variant code (UAR for Gln) independently, such as dasycladacean green algae (e.g. Acetabularia) and diplomonads (e.g. Spironucleus), should provide important clues to understanding eRF1 codon recognition.
APPENDIX
Recently two eRF1 genes of Euplotes octocarinatus became available in the GenBank database (AF245454 and AJ272501). Our preliminary phylogenetic analysis clearly indicated that the E.octocarinatus eRF1a (AJ272501) and E.aediculatus eRF1A (AF29881) and E.octocarinatus eRF1b (AF245454) and E.aediculatus eRF1B (AF29882) are orthologous.
Acknowledgments
ACKNOWLEDGEMENTS
We thank S.Douglas (Institute for Marine Bioscience, Halifax, Canada) and S.DasSarma (University of Massachusetts, Amherst, MA) for providing Guillardia nucleomorph eRF1 and Halobacterium sp. NRC-1 aRF1 sequences, respectively, before publication. D.M.Prescott (University of Colorado, Boulder, UT) generously provided the macronuclear DNA samples of O.trifallax and E.aediculatus and rechecked the DNA sequence AF188150 at our request. The M.mazei aRF1 sequence and a D.discoideum cDNA clone (SLI621) were kindly provided by the Göttingen Genomic Laboratory (University of Göttingen, Göttingen, Germany) and the Dictyostelium cDNA Project (Tsukuba University, Tsukuba, Japan), respectively. We also thank members of the Doolittle and Roger laboratories for helpful discussions and critical review of this manuscript. Y.I. was supported by a post-doctoral research fellowship from the Japanese Society for the Promotion of Science for Young Scientists Abroad. This work was supported by grant MT4467 from the Canadian Medical Research Council to W.F.D.
DDBJ/EMBL/GenBank accession nos AF298830–AF298834
References
- 1.Osawa S., Jukes,T.H., Watanabe,K. and Muto,A. (1992) Recent evidence for evolution of the genetic code. Microbiol. Rev., 56, 229–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckingham R.H., Grentzmann,G. and Kisselev,L. (1997) Polypeptide chain release factors. Mol. Microbiol., 24, 449–456. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura Y. and Ito,K. (1998) How protein reads the stop codon and terminates translation. Genes Cells, 3, 265–278. [DOI] [PubMed] [Google Scholar]
- 4.Inagaki Y., Bessho,Y. and Osawa,S. (1993) Lack of peptide-release activity responding to codon UGA in Mycoplasma capricolum. Nucleic Acids Res., 21, 1335–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser C.M., Gocayne,J.D., White,O., Adams,M.D., Clayton,R.A., Fleischmann,R.D., Bult,C.J., Kerlavage,A.R., Sutton,G., Kelley,J.M. et al. (1995) The minimal gene complement of Mycoplasma genitalium. Science, 270, 397–403. [DOI] [PubMed] [Google Scholar]
- 6.Himmelreich R., Hilbert,H., Plagens,H., Pirkl,E., Li,B.C. and Herrmann,R. (1996) Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res., 24, 4420–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frolova L., Le Goff,X., Rasmussen,H.H., Cheperegin,S., Drugeon,G., Kress,M., Arman,I., Haenni,A.L., Celis,J.E., Philippe,M., Justesen,J. and Kisselev,L. (1994) A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature, 372, 701–703. [DOI] [PubMed] [Google Scholar]
- 8.Dontsova M., Frolova,L., Vassilieva,J., Piendl,W., Kisselev,L. and Garber,M. (2000) Translation termination factor aRF1 from the archaeon Methanococcus jannaschii is active with eukaryotic ribosomes. FEBS Lett., 472, 213–216. [DOI] [PubMed] [Google Scholar]
- 9.Ito K., Ebihara,K., Uno,M. and Nakamura,Y. (1996) Conserved motifs in prokaryotic and eukaryotic polypeptide release factors: tRNA-protein mimicry hypothesis. Proc. Natl Acad. Sci. USA, 93, 5443–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito K., Uno,M. and Nakamura,Y. (2000) A tripeptide ‘anticodon’ deciphers stop codons in messenger RNA. Nature, 403, 680–684. [DOI] [PubMed] [Google Scholar]
- 11.Karamyshev A.L., Ito,K. and Nakamura,Y. (1999) Polypeptide release factor eRF1 from Tetrahymena thermophila: cDNA cloning, purification and complex formation with yeast eRF3. FEBS Lett., 457, 483–488. [DOI] [PubMed] [Google Scholar]
- 12.Song H., Mugnier,P., Das,A.K., Webb,H.M., Evans,D.R., Tuite,M.F., Hemmings,B.A. and Barford,D. (2000) The crystal structure of human eukaryotic release factor eRF1—mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell, 100, 311–321. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura Y., Ito,K. and Ehrenberg,M. (2000) Mimicry grasps reality in translation termination. Cell, 101, 349–352. [DOI] [PubMed] [Google Scholar]
- 14.Knight R.D. and Landweber,L.F. (2000) The early evolution of the genetic code. Cell, 101, 569–572. [DOI] [PubMed] [Google Scholar]
- 15.Tourancheau A.B., Tsao,N., Klobutcher,L.A., Pearlman,R.E. and Adoutte,A. (1995) Genetic code deviations in the ciliates: evidence for multiple and independent events. EMBO J., 14, 3262–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prescott D.M. (1994) The DNA of ciliated protozoa. Microbiol. Rev., 68, 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morio T., Urushihara,H., Saito,T., Ugawa,Y., Mizuno,H., Yoshida,M., Yoshino,R., Mitra,B.N., Pi,M., Sato,T. et al. (1998) The Dictyostelium developmental cDNA project: generation and analysis of expressed sequence tags from the first-finger stage of development. DNA Res., 5, 335–340. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strimmer K. and von Haeseler,A. (1996) Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol. Biol. Evol., 13, 964–969. [Google Scholar]
- 20.Felsenstein J. (1993) PHYLIP Version 3.57c Ed. University of Washington, Seattle, WA.
- 21.Inagaki Y. and Doolittle,W.F. (2000) Evolution of the eukaryotic translation termination system: origins of release factors. Mol. Biol. Evol., 17, 882–889. [DOI] [PubMed] [Google Scholar]
- 22.Sogin M.L. (1997) History assignment: when was the mitochondrion founded? Curr. Opin. Genet. Dev., 7, 792–799. [DOI] [PubMed] [Google Scholar]
- 23.Cavalier-Smith T. (1993) Kingdom protozoa and its 18 phyla. Microbiol. Rev., 57, 953–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert W. and Glynias,M. (1993) On the ancient nature of introns. Gene, 135, 137–144. [DOI] [PubMed] [Google Scholar]
- 25.Stoltzfus A., Logsdon,J.M.,Jr, Palmer,J.D. and Doolittle,W.F. (1997) Intron ‘sliding’ and the diversity of intron positions. Proc. Natl Acad. Sci. USA, 94, 10739–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito K., Ebihara,K. and Nakamura,Y. (1998) The stretch of C-terminal acidic amino acids of translational release factor eRF1 is a primary binding site for eRF3 of fission yeast. RNA, 4, 958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebihara K. and Nakamura,Y. (1999) C-terminal interaction of translational release factors eRF1 and eRF3 of fission yeast: G-domain uncoupled binding and the role of conserved amino acids. RNA, 5, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eurwilaichitr L., Graves,F.M., Stansfield,I. and Tuite,M.F. (1999) The C-terminus of eRF1 defines a functionally important domain for translation termination in Saccharomyces cerevisiae. Mol. Microbiol., 32, 485–496. [DOI] [PubMed] [Google Scholar]
- 29.Frolova L.Y., Merkulova,T.I. and Kisselev,L.L. (2000) Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA, 6, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Douglas S.E. and Penny,S.L. (1999) The plastid genome of the cryptophyte alga, Guillardia theta: complete sequence and conserved synteny groups confirm its common ancestry with red algae. J. Mol. Evol., 48, 236–244. [DOI] [PubMed] [Google Scholar]
- 31.Frolova L.Y., Tsivkovskii,R.Y., Sivolobova,G.F., Oparina,N.Y., Serpinsky,O.I., Blinov,V.M., Tatkov,S.I. and Kisselev,L.L. (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA, 5, 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertram G., Bell,H.A., Ritchie,D.W., Fullerton,G. and Stansfield,I. (2000) Terminationg eukaryotic translation: domain 1 of release factor eRF1 functions in stop codon recongnition. RNA, 6, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]