

Abstract
In the crystal structure of the title compound, C9H9NO3S, there is distorted tetrahedral geometry around the S atom. The sulfonyl group is almost normal to the benzene ring, while the carbonyl O atom and methyl C atom are on opposite sides of this ring. The heterocyclic ring adopts a half-boat conformation with the S atom out of the plane. The molecules are dimerized by hydrogen bonding involving the benzene ring and the sulfonyl group. These dimers are linked to each other in the same way. There is an intramolecular hydrogen bond between a methyl C—H group and a sulfonyl O atom, and a π–π interaction between the aromatic rings of two dimers at a centroid-to-centroid distance of 3.6373 (13) Å.
Related literature
For related literature, see: Cremer & Pople (1975 ▶); Harmata et al. (2004 ▶); Lombardino (1972 ▶); Misu & Togo (2003 ▶); Shafiq et al. (2008 ▶); Siddiqui et al. (2007 ▶).
Experimental
Crystal data
C9H9NO3S
M r = 211.23
Triclinic,
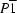
a = 7.4553 (4) Å
b = 8.5437 (4) Å
c = 8.7097 (4) Å
α = 67.691 (2)°
β = 70.467 (2)°
γ = 66.327 (2)°
V = 459.09 (4) Å3
Z = 2
Mo Kα radiation
μ = 0.33 mm−1
T = 296 (2) K
0.20 × 0.15 × 0.10 mm
Data collection
Bruker Kappa APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2007 ▶) T min = 0.952, T max = 0.971
7205 measured reflections
1787 independent reflections
1678 reflections with I > 2σ(I)
R int = 0.019
Refinement
R[F 2 > 2σ(F 2)] = 0.032
wR(F 2) = 0.095
S = 1.09
1678 reflections
127 parameters
H-atom parameters constrained
Δρmax = 0.22 e Å−3
Δρmin = −0.42 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: APEX2; data reduction: SAINT (Bruker, 2007 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶) and PLATON (Spek, 2003 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶) and PLATON.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808003498/bq2063sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808003498/bq2063Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯O1i | 0.93 | 2.58 | 3.432 (2) | 152 |
| C4—H4⋯O2ii | 0.93 | 2.46 | 3.238 (3) | 142 |
| C9—H9A⋯O2 | 0.96 | 2.31 | 2.830 (3) | 113 |
Symmetry codes: (i) 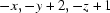 ; (ii)
; (ii) 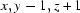 .
.
Acknowledgments
The authors acknowledge the Higher Education Commission, Islamabad, Pakistan, and Bana International, Karachi, Pakistan, for funding the purchase of the diffractometer and for technical support, respectively.
supplementary crystallographic information
Comment
Benzothiazines belong to an important heterocyclic class of compounds which find a number of applications in medicinal chemistry. Derivatives of 2,1-benzothiazines were reported to possess potent biological activities such as lipoxygenase inhibition and are used as drugs for heart diseases (Misu, & Togo, 2003). These are used as intermediate precursors for the preparation of drugs used for curing Tuberculosis (Harmata et al., 2004). The importance of 2,1-benzothiazine derivatives in medicinal chemistry has brought enormous attention to their synthesis. Herein, we report the crystal structure of the title compound.
The structure determination of the title compound (I) is in continuation to our research on derivatives of 2,1 benzothiazine (Shafiq et al., 2008). The structural isomer (II), 2-methyl-2H-1,2-benzothiazin-4(3H)one 1,1-dioxide of (I) has been reported (Siddiqui et al., 2007).
In the title compound the bond distances S1—N1 [1.6429 (13) Å] and S1—C8 [1.7514 (17) Å], have been significantly changed in comparison to 1.629 (3) Å and 1.760 (4) Å respectively as reported in (II). The change in bond angles around S is large enough which is evident from the range [99.47 (8)°-118.32 (8)°] in (I), as compared to (II) [103.05 (16)°- 118.66 (16)°]. All the atoms in heterocyclic thiazine as well as C-atoms of phenyl ring are nearly planer except that of S1. The S1 is displaced by 0.7834 (15) Å from the plane (a) defined by (C1 to C8, N1) and C-atom of methyl group is at a distance of 0.340 (3) Å from it. Thus the atoms of the two rings form a long half boat confirmation. The plane (b) defined by the atoms (O1,S1,O2) makes a dihedral angle of 89.81 (6)° to the plane (a) and hence these two planes are almost normal to each other. The O1-atom is at longest distance of -2.1912 (16) Å from plane (a). In the asymmetric unit there is an intramolecular H-bond as given in Table 2. The confirmation of the hetrocyclic ring in terms of the puckering parameters (Cremer & Pople, 1975) is described by; Q = 0.5767 (14) Å, θ = 54.98 (16)° and φ = 359.7 (2)°. The title compound is basically dimerized by H-bonding through C2—H2···O1i (i = -x, -y + 2, -z + 1) as shown in Fig 2. It is interesting that the ring formed in dimer is of twelve bonds to which the methyl groups adopt cis, trans position. The dimers are linked to each other by symmetry code ii = x, y - 1, z + 1. The closest interaction [3.283 (2) Å] occurs between O1···O1iii (symmetry code: iii = -x, -y + 2, -z), other than atoms involved in intermolecular H-bond. There is no X—H···Cg bond, however there exist a π-π interaction between the aromatic rings of two dimers at a distance of 3.6373 (13) Å by the symmetry operation 1 - x, 1 - y, 1 - z.
Experimental
The title compound (I) was synthesized using the reported procedure (Lombardino, 1972) and crystals were grown by slow evaporation from a solution of CH3OH at 298 K.
Figures
Fig. 1.

ORTEP drawing of the title compound, C10H11NO3S, with the atom numbering scheme. The thermal ellipsoids are drawn at the 50% probability level.
Fig. 2.

The packing figure (PLATON: Spek, 2003) which shows the dimeric nature of the compound owing to inter molecular hydrogen bondings and also showing a link between dimers.
Crystal data
| C9H9NO3S | Z = 2 |
| Mr = 211.23 | F000 = 220 |
| Triclinic, P1 | Dx = 1.528 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation radiation λ = 0.71073 Å |
| a = 7.4553 (4) Å | Cell parameters from 1678 reflections |
| b = 8.5437 (4) Å | θ = 2.6–25.9º |
| c = 8.7097 (4) Å | µ = 0.33 mm−1 |
| α = 67.691 (2)º | T = 296 (2) K |
| β = 70.467 (2)º | Prismatic, colourless |
| γ = 66.327 (2)º | 0.20 × 0.15 × 0.10 mm |
| V = 459.09 (4) Å3 |
Data collection
| Bruker Kappa-APEXII CCD diffractometer | 1787 independent reflections |
| Radiation source: fine-focus sealed tube | 1678 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.020 |
| Detector resolution: 7.50 pixels mm-1 | θmax = 25.9º |
| T = 296(2) K | θmin = 2.6º |
| ω scans | h = −9→9 |
| Absorption correction: multi-scan(SADABS; Bruker, 2007) | k = −10→10 |
| Tmin = 0.952, Tmax = 0.971 | l = −10→10 |
| 7205 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.032 | H-atom parameters constrained |
| wR(F2) = 0.095 | w = 1/[σ2(Fo2) + (0.0531P)2 + 0.1411P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.09 | (Δ/σ)max < 0.001 |
| 1678 reflections | Δρmax = 0.22 e Å−3 |
| 127 parameters | Δρmin = −0.42 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.18773 (6) | 0.94371 (5) | 0.15987 (5) | 0.03811 (16) | |
| O1 | −0.02000 (18) | 0.95817 (16) | 0.20416 (15) | 0.0472 (3) | |
| O2 | 0.2454 (2) | 1.09673 (18) | 0.05037 (17) | 0.0618 (4) | |
| O3 | 0.3153 (2) | 0.46358 (19) | 0.16958 (19) | 0.0641 (4) | |
| N1 | 0.2759 (2) | 0.87608 (18) | 0.33158 (17) | 0.0410 (3) | |
| C1 | 0.2522 (2) | 0.71412 (19) | 0.45108 (18) | 0.0327 (3) | |
| C2 | 0.2208 (3) | 0.6907 (2) | 0.6241 (2) | 0.0447 (4) | |
| H2 | 0.2151 | 0.7814 | 0.6616 | 0.054* | |
| C3 | 0.1982 (3) | 0.5318 (3) | 0.7405 (2) | 0.0540 (5) | |
| H3 | 0.1782 | 0.5166 | 0.8559 | 0.065* | |
| C4 | 0.2048 (3) | 0.3964 (3) | 0.6881 (2) | 0.0548 (5) | |
| H4 | 0.1883 | 0.2908 | 0.7672 | 0.066* | |
| C5 | 0.2359 (2) | 0.4185 (2) | 0.5186 (2) | 0.0462 (4) | |
| H5 | 0.2398 | 0.3270 | 0.4833 | 0.055* | |
| C6 | 0.2618 (2) | 0.57543 (19) | 0.39688 (19) | 0.0341 (3) | |
| C7 | 0.2997 (2) | 0.5863 (2) | 0.2155 (2) | 0.0399 (4) | |
| C8 | 0.3241 (3) | 0.7571 (2) | 0.0823 (2) | 0.0434 (4) | |
| H8A | 0.2785 | 0.7728 | −0.0162 | 0.052* | |
| H8B | 0.4650 | 0.7481 | 0.0465 | 0.052* | |
| C9 | 0.3194 (3) | 1.0018 (3) | 0.3773 (3) | 0.0555 (5) | |
| H9A | 0.3301 | 1.1026 | 0.2801 | 0.083* | |
| H9B | 0.4436 | 0.9450 | 0.4149 | 0.083* | |
| H9C | 0.2133 | 1.0409 | 0.4671 | 0.083* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0511 (3) | 0.0305 (2) | 0.0313 (2) | −0.01201 (17) | −0.01384 (17) | −0.00395 (16) |
| O1 | 0.0486 (7) | 0.0447 (7) | 0.0460 (7) | −0.0040 (5) | −0.0179 (5) | −0.0152 (5) |
| O2 | 0.0921 (11) | 0.0419 (7) | 0.0480 (7) | −0.0301 (7) | −0.0247 (7) | 0.0087 (6) |
| O3 | 0.0813 (10) | 0.0563 (8) | 0.0664 (9) | −0.0287 (7) | 0.0007 (7) | −0.0364 (7) |
| N1 | 0.0614 (9) | 0.0327 (7) | 0.0371 (7) | −0.0211 (6) | −0.0205 (6) | −0.0033 (6) |
| C1 | 0.0317 (7) | 0.0311 (7) | 0.0322 (7) | −0.0082 (6) | −0.0092 (6) | −0.0055 (6) |
| C2 | 0.0462 (9) | 0.0493 (10) | 0.0356 (8) | −0.0100 (7) | −0.0121 (7) | −0.0113 (7) |
| C3 | 0.0454 (10) | 0.0683 (13) | 0.0313 (8) | −0.0154 (9) | −0.0091 (7) | 0.0017 (8) |
| C4 | 0.0474 (10) | 0.0477 (10) | 0.0511 (11) | −0.0196 (8) | −0.0126 (8) | 0.0116 (8) |
| C5 | 0.0414 (9) | 0.0330 (8) | 0.0580 (11) | −0.0132 (7) | −0.0129 (8) | −0.0029 (7) |
| C6 | 0.0304 (7) | 0.0293 (7) | 0.0386 (8) | −0.0084 (6) | −0.0068 (6) | −0.0072 (6) |
| C7 | 0.0364 (8) | 0.0391 (8) | 0.0458 (9) | −0.0101 (6) | −0.0046 (6) | −0.0193 (7) |
| C8 | 0.0500 (9) | 0.0451 (9) | 0.0317 (8) | −0.0113 (7) | −0.0055 (7) | −0.0141 (7) |
| C9 | 0.0770 (13) | 0.0443 (10) | 0.0624 (11) | −0.0274 (9) | −0.0267 (10) | −0.0143 (8) |
Geometric parameters (Å, °)
| S1—O2 | 1.4262 (13) | C3—H3 | 0.9300 |
| S1—O1 | 1.4302 (13) | C4—C5 | 1.365 (3) |
| S1—N1 | 1.6429 (13) | C4—H4 | 0.9300 |
| S1—C8 | 1.7574 (17) | C5—C6 | 1.397 (2) |
| O3—C7 | 1.209 (2) | C5—H5 | 0.9300 |
| N1—C1 | 1.4175 (18) | C6—C7 | 1.482 (2) |
| N1—C9 | 1.452 (2) | C7—C8 | 1.515 (2) |
| C1—C2 | 1.393 (2) | C8—H8A | 0.9700 |
| C1—C6 | 1.402 (2) | C8—H8B | 0.9700 |
| C2—C3 | 1.387 (2) | C9—H9A | 0.9600 |
| C2—H2 | 0.9300 | C9—H9B | 0.9600 |
| C3—C4 | 1.375 (3) | C9—H9C | 0.9600 |
| O2—S1—O1 | 118.32 (8) | C4—C5—C6 | 121.56 (17) |
| O2—S1—N1 | 107.31 (8) | C4—C5—H5 | 119.2 |
| O1—S1—N1 | 110.55 (7) | C6—C5—H5 | 119.2 |
| O2—S1—C8 | 111.61 (9) | C5—C6—C1 | 118.92 (15) |
| O1—S1—C8 | 107.95 (8) | C5—C6—C7 | 117.93 (14) |
| N1—S1—C8 | 99.47 (8) | C1—C6—C7 | 123.14 (14) |
| C1—N1—C9 | 121.70 (14) | O3—C7—C6 | 122.71 (16) |
| C1—N1—S1 | 116.58 (10) | O3—C7—C8 | 118.80 (15) |
| C9—N1—S1 | 119.32 (12) | C6—C7—C8 | 118.48 (13) |
| C2—C1—C6 | 119.27 (14) | C7—C8—S1 | 111.72 (11) |
| C2—C1—N1 | 120.09 (14) | C7—C8—H8A | 109.3 |
| C6—C1—N1 | 120.63 (13) | S1—C8—H8A | 109.3 |
| C3—C2—C1 | 119.87 (17) | C7—C8—H8B | 109.3 |
| C3—C2—H2 | 120.1 | S1—C8—H8B | 109.3 |
| C1—C2—H2 | 120.1 | H8A—C8—H8B | 107.9 |
| C4—C3—C2 | 121.05 (17) | N1—C9—H9A | 109.5 |
| C4—C3—H3 | 119.5 | N1—C9—H9B | 109.5 |
| C2—C3—H3 | 119.5 | H9A—C9—H9B | 109.5 |
| C5—C4—C3 | 119.31 (16) | N1—C9—H9C | 109.5 |
| C5—C4—H4 | 120.3 | H9A—C9—H9C | 109.5 |
| C3—C4—H4 | 120.3 | H9B—C9—H9C | 109.5 |
| O2—S1—N1—C1 | −174.04 (12) | C4—C5—C6—C1 | −1.1 (2) |
| O1—S1—N1—C1 | 55.60 (13) | C4—C5—C6—C7 | 178.55 (15) |
| C8—S1—N1—C1 | −57.74 (13) | C2—C1—C6—C5 | 1.2 (2) |
| O2—S1—N1—C9 | 23.23 (18) | N1—C1—C6—C5 | −179.41 (14) |
| O1—S1—N1—C9 | −107.13 (15) | C2—C1—C6—C7 | −178.43 (14) |
| C8—S1—N1—C9 | 139.53 (15) | N1—C1—C6—C7 | 0.9 (2) |
| C9—N1—C1—C2 | 15.9 (2) | C5—C6—C7—O3 | −2.5 (2) |
| S1—N1—C1—C2 | −146.39 (13) | C1—C6—C7—O3 | 177.15 (16) |
| C9—N1—C1—C6 | −163.48 (16) | C5—C6—C7—C8 | 178.83 (14) |
| S1—N1—C1—C6 | 34.24 (18) | C1—C6—C7—C8 | −1.5 (2) |
| C6—C1—C2—C3 | −0.5 (2) | O3—C7—C8—S1 | 151.98 (15) |
| N1—C1—C2—C3 | −179.84 (14) | C6—C7—C8—S1 | −29.30 (18) |
| C1—C2—C3—C4 | −0.4 (3) | O2—S1—C8—C7 | 166.75 (12) |
| C2—C3—C4—C5 | 0.6 (3) | O1—S1—C8—C7 | −61.58 (13) |
| C3—C4—C5—C6 | 0.2 (3) | N1—S1—C8—C7 | 53.77 (13) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O1i | 0.93 | 2.58 | 3.432 (2) | 152 |
| C4—H4···O2ii | 0.93 | 2.46 | 3.238 (3) | 142 |
| C9—H9A···O2 | 0.96 | 2.31 | 2.830 (3) | 113 |
Symmetry codes: (i) −x, −y+2, −z+1; (ii) x, y−1, z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BQ2063).
References
- Bruker (2007). APEX2 (Version 1.27), SAINT (Version 7.12a) and SADABS (Version 2004/I). Bruker AXS Inc., Madison, Wisconsin, USA.
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc.97, 1354–1358.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Harmata, M., Hong, X. & Barnes, C. L. (2004). Org. Lett.6, 2201–2203. [DOI] [PubMed]
- Lombardino, J. G. (1972). J. Heterocycl. Chem.9, 315–317.
- Misu, Y. & Togo, H. (2003). Org. Biomol. Chem.1, 1342–1346. [DOI] [PubMed]
- Shafiq, M., Tahir, M. N., Khan, I. U., Siddiqui, W. A. & Arshad, M. N. (2008). Acta Cryst. E64, o389. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siddiqui, W. A., Ahmad, S., Siddiqui, H. L., Tariq, M. I. & Parvez, M. (2007). Acta Cryst. E63, o4585.
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808003498/bq2063sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808003498/bq2063Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report