

Abstract
The asymmetric unit of the title compound, C8H6N2O4, contains one half-molecule; a twofold rotation axis bisects the molecule. The quinoxaline ring is planar, which can be attributed to electron delocalization. In the crystal structure, intermolecular O—H⋯O hydrogen bonds link the molecules into R 2 2(10) motifs, leading to layers, which interact via phenyl–phenyl interactions (C⋯C distances in the range 3.238–3.521 Å).
Related literature
For general background, see: Zarranz et al. (2004 ▶); Chowdhury et al. (2004 ▶); Monge et al. (1995 ▶); Fuchs et al. (2001 ▶); Dance (1996 ▶); Bernstein et al. (1995 ▶). For related literature, see: Elina & Tsyrul’nikova (1963 ▶); Akkurt et al. (2004 ▶); Mustaphi et al. (2001 ▶); Oxtoby et al. (2005 ▶); Ley & Seng (1975 ▶); For bond-length data, see: Allen et al. (1987 ▶);
Experimental
Crystal data
C8H6N2O4
M r = 194.15
Orthorhombic,
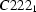
a = 4.2562 (6) Å
b = 17.630 (3) Å
c = 10.4775 (17) Å
V = 786.2 (2) Å3
Z = 4
Mo Kα radiation
μ = 0.14 mm−1
T = 294 (2) K
0.50 × 0.20 × 0.10 mm
Data collection
Nicolet P3 diffractometer
Absorption correction: none
1004 measured reflections
529 independent reflections
437 reflections with I > 2σ(I)
R int = 0.022
3 standard reflections every 50 reflections intensity decay: 2%
Refinement
R[F 2 > 2σ(F 2)] = 0.034
wR(F 2) = 0.071
S = 1.07
529 reflections
69 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.12 e Å−3
Δρmin = −0.15 e Å−3
Data collection: P3/PC Data Collection Software (Siemens, 1991 ▶); cell refinement: P3/PC Data Collection Software; data reduction: SHELXTL-Plus (Sheldrick, 2008 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL-Plus; software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2003 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808003784/hk2425sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808003784/hk2425Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯O2i | 0.96 (3) | 1.63 (3) | 2.584 (2) | 174 (3) |
Symmetry code: (i) 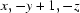 .
.
Acknowledgments
We acknowledge financial support from Al al-Bayt University (Jordan). We are grateful for a research grant from Deutsche Forschungsgemeinschaft (DFG) 2007 (to R. Abu-El-Halawa) and for the generous hospitality and discussions of Prof Volker Jäger and to Helmut Griesser at the Institute of Organic Chemistry, University of Stuttgart, Germany. We also thank Mr Raed Soudqi for his help.
supplementary crystallographic information
Comment
Quinoxalines are of interest owing to their biological activities. They seem to have very interesting anticancer activity (Zarranz et al., 2004). For example, 3-aminoquinoxaline-2-carbonitrile 1,4-dioxide have been studied extensively as bioreductive cytotoxic agent. It was found to be an efficient agent and causes redox-activated DNA damage (Chowdhury et al., 2004), even more active than the first drug clinically used as bioreductive cytotoxic agent (Monge et al., 1995; Fuchs et al., 2001). A nonconvenient synthesis of the title compound, (I), was reported previously (Elina & Tsyruľ nikova, 1963), via the hydrolysis of 2-amino-3-hydroxyquinoxaline 1,4-dioxide, which results as a side product (4%) from oxidation of 2-acetamidoquinoxaline with acetic peroxide acid using boiling HCl solution. We report herein a novel simple synthetic method for (I), along with its crystal structure.
The new synthetic strategy for (I), (Fig. 1), is based on the reaction of (1) with NaOH solution, yielding (2) in an SNAr reaction, which upon hydrolysis with boiling HCl solution, via protonation of amine followed by the attack of water molecule, yielded (I) in a good amount (90%).
The asymmetric unit of the title compound, (I), (Fig. 2) contains one half-molecule. The quinoxaline ring is planar, which can be attributed to a wide range of electron delocalization. Bond lengths and angles are in accordance with the corresponding reported values in 1,4-dihydroquinoxaline -2,3-dione core (Oxtoby et al., 2005) and other similar N-alkyl quinoxalines (Akkurt et al., 2004; Mustaphi et al., 2001). The existence of (I) in the dione form is evident from C1—O2 [1.226 (3) Å] bond, being smaller than a pure single bond, which confirms the double bond character (Allen et al., 1987). The C1—C1i [1.503 (4) Å] bond has single bond character compared to multiple bond characters in C2—C2i [1.381 (4) Å], C2—C3 [1.391 (3) Å], C3—C4 [1.382 (3) Å] and C4—C4i [1.372 (6) Å] [symmetry code: (i) -x, y, 1/2 - z]. The N1—C1 [1.345 (3) Å] bond is significantly shorter than N1—C2 [1.404 (3) Å] and it is an intermediate between those typical for the corresponding single and double bonds, suggesting some degree of delocalization. The N1—C1 bond length is closer to the average Car—Nsp2 (planar) value of 1.353 (7) Å rather than the Car—Nsp3 (pyramidal) value of 1.419 (17) Å (Allen et al., 1987), with the sum of the bond angles around atom N1 [359.81 (18)°], indicating sp2 hybridization.
In the crystal structure, intermolecular O—H···O hydrogen bonds (Table 1) link the molecules into R22(10) motifs (Fig. 3) (Bernstein et al., 1995) leading to layers running along the c axis (Fig. 4). Molecules within layers are further interacting via phenyl···phenyl interactions (Dance, 1996), where the layers parallel to a axis interact in an offset stacking motif (C···C distances in the range of 3.238–3.521 Å).
Experimental
For the preparation of (I), to a suspension solution of (1) (2.02 g, 10 mmol) (Ley & Seng, 1975) in ethanol (20 ml), NaOH (20 ml, 10%) was added to give a deep blue solution. After refluxing for 5 h, the brown solution was allowed to cool to room temperature. The resulting mixture was then treated with HCl (30 ml, 10%), refluxed for another 5 h and then allowed to stand undisturbed. The resulting residual brown solid was filtered off, washed with cold water (5 ml) and then by cold ethanol (5 ml). The title compound, (I), was recrystallized from ethanol solution (yield; 1.76 g, 90%, m.p. 535–536 K decomposition). Analysis found: C 49.45, H 3.27, N 14.41%; C8H6N2O4 requires: C 49.49, H 3.12, N 14.43%. 1H NMR (300 MHz, DMSO-d6): δ = 7.36 (m, 2H; H4/H7), 7.56 (m, 2H; H5/H6); 13C NMR (75 MHz, DMSO-d6): δ = 111.6 (C4/C7), 123.3 (C5/C6), 124.0 (C4a/C7a), 150.4 (C2/C3) p.p.m.. ESI: m/z = 217.02 (C8H6N2O4Na).
Refinement
H atom (for OH) was located in a difference synthesis and refined isotropically [O—H = 0.96 (3) Å; Uiso(H) = 0.072 (10) Å2]. The remaining H atoms were positioned geometrically, with C—H = 0.93 Å for aromatic H and constrained to ride on their parent atoms, with Uiso(H) = 1.2Ueq(C).
Figures
Fig. 1.

Schematic representation for the steps through which reaction proceeds.
Fig. 2.

The molecular structure of the title molecule, with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 3.

Part of the crystal structure of (I), showing the formation of R22(10) motifs.
Fig. 4.

A packing diagram of (I), showing the layers of molecules parallel to c axis. All hydrogen atoms were omitted for clarity. Hydrogen bonds are shown as dashed lines [symmetry codes: (i) -x, y, -z + 1/2, (ii) x, -y + 1, -z].
Crystal data
| C8H6N2O4 | F000 = 400 |
| Mr = 194.15 | Dx = 1.640 Mg m−3 |
| Orthorhombic, C2221 | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: C 2c 2 | Cell parameters from 20 reflections |
| a = 4.2562 (6) Å | θ = 14–16º |
| b = 17.630 (3) Å | µ = 0.14 mm−1 |
| c = 10.4775 (17) Å | T = 294 (2) K |
| V = 786.2 (2) Å3 | Plates, colourless |
| Z = 4 | 0.50 × 0.20 × 0.10 mm |
Data collection
| Nicolet P3 diffractometer | Rint = 0.022 |
| Radiation source: fine-focus sealed tube | θmax = 27.0º |
| Monochromator: graphite | θmin = 2.3º |
| T = 294(2) K | h = 0→5 |
| Wyckoff scan | k = 0→22 |
| Absorption correction: none | l = −13→13 |
| 1004 measured reflections | 3 standard reflections |
| 529 independent reflections | every 50 reflections |
| 437 reflections with I > 2σ(I) | intensity decay: 2% |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.034 | w = 1/[σ2(Fo2) + (0.0208P)2 + 0.4073P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.071 | (Δ/σ)max < 0.001 |
| S = 1.07 | Δρmax = 0.12 e Å−3 |
| 529 reflections | Δρmin = −0.15 e Å−3 |
| 69 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.013 (2) |
| Secondary atom site location: difference Fourier map |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.2116 (4) | 0.41727 (11) | 0.15388 (16) | 0.0350 (5) | |
| C1 | 0.1130 (6) | 0.48556 (11) | 0.1948 (2) | 0.0364 (6) | |
| O1 | 0.4527 (4) | 0.41624 (10) | 0.06569 (15) | 0.0456 (5) | |
| H1 | 0.357 (7) | 0.4338 (15) | −0.012 (3) | 0.072 (10)* | |
| C2 | 0.1068 (5) | 0.34702 (11) | 0.2004 (2) | 0.0332 (5) | |
| O2 | 0.1965 (5) | 0.54622 (9) | 0.14878 (15) | 0.0531 (6) | |
| C3 | 0.2131 (7) | 0.27901 (13) | 0.1487 (2) | 0.0456 (7) | |
| H3 | 0.3541 | 0.2788 | 0.0808 | 0.055* | |
| C4 | 0.1046 (7) | 0.21177 (13) | 0.2002 (2) | 0.0557 (8) | |
| H4 | 0.1745 | 0.1659 | 0.1669 | 0.067* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0369 (10) | 0.0401 (9) | 0.0279 (8) | 0.0022 (10) | 0.0011 (9) | −0.0011 (8) |
| C1 | 0.0452 (15) | 0.0371 (12) | 0.0269 (10) | −0.0036 (11) | −0.0014 (13) | −0.0019 (9) |
| O1 | 0.0416 (9) | 0.0631 (10) | 0.0320 (8) | 0.0113 (10) | 0.0045 (9) | 0.0052 (9) |
| C2 | 0.0354 (14) | 0.0334 (10) | 0.0309 (10) | 0.0004 (10) | −0.0096 (12) | −0.0007 (8) |
| O2 | 0.0823 (16) | 0.0374 (8) | 0.0397 (9) | −0.0123 (10) | 0.0136 (13) | 0.0017 (7) |
| C3 | 0.0519 (16) | 0.0435 (13) | 0.0415 (13) | 0.0099 (13) | −0.0118 (16) | −0.0077 (10) |
| C4 | 0.072 (2) | 0.0336 (11) | 0.0615 (16) | 0.0086 (13) | −0.0242 (18) | −0.0079 (11) |
Geometric parameters (Å, °)
| N1—C1 | 1.345 (3) | C2—C2i | 1.381 (4) |
| N1—O1 | 1.381 (2) | C2—C3 | 1.391 (3) |
| N1—C2 | 1.404 (3) | C3—C4 | 1.382 (3) |
| C1—O2 | 1.226 (3) | C3—H3 | 0.9300 |
| C1—C1i | 1.503 (4) | C4—C4i | 1.372 (6) |
| O1—H1 | 0.96 (3) | C4—H4 | 0.9300 |
| C1—N1—O1 | 117.21 (19) | C2i—C2—N1 | 118.08 (11) |
| C1—N1—C2 | 125.42 (18) | C3—C2—N1 | 121.5 (2) |
| O1—N1—C2 | 117.18 (18) | C4—C3—C2 | 118.6 (2) |
| O2—C1—N1 | 124.4 (2) | C4—C3—H3 | 120.7 |
| O2—C1—C1i | 119.22 (14) | C2—C3—H3 | 120.7 |
| N1—C1—C1i | 116.41 (12) | C4i—C4—C3 | 120.93 (16) |
| N1—O1—H1 | 104.2 (17) | C4i—C4—H4 | 119.5 |
| C2i—C2—C3 | 120.47 (15) | C3—C4—H4 | 119.5 |
| O1—N1—C1—O2 | 8.8 (3) | C1—N1—C2—C3 | 177.4 (2) |
| C2—N1—C1—O2 | −176.4 (2) | O1—N1—C2—C3 | −7.8 (3) |
| O1—N1—C1—C1i | −170.8 (2) | C2i—C2—C3—C4 | −1.1 (4) |
| C2—N1—C1—C1i | 4.1 (4) | N1—C2—C3—C4 | 178.9 (2) |
| C1—N1—C2—C2i | −2.5 (4) | C2—C3—C4—C4i | 0.3 (5) |
| O1—N1—C2—C2i | 172.3 (2) |
Symmetry codes: (i) −x, y, −z+1/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2ii | 0.96 (3) | 1.63 (3) | 2.584 (2) | 174 (3) |
Symmetry codes: (ii) x, −y+1, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2425).
References
- Akkurt, M., Öztürk, S., Küçükbay, H., Orhan, E. & Büyükgüngör, O. (2004). Acta Cryst. E60, o1266–o1268.
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans.2, pp. S1–19.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Chowdhury, G., Kotandeniya, D., Daniels, J. S., Barnes, C. L. & Gates, K. S. (2004). Chem. Res. Toxicol.17, 1399–1405. [DOI] [PubMed]
- Dance, I. G. (1996). Supramolecular Inorganic Chemistry, in The Crystal as a Supramolecular Entity, edited by G. R. Desiraju, pp. 137–233. New York: John Wiley.
- Elina, A. S. & Tsyrul’nikova, L. G. (1963). Zh. Obshch. Khim.33, 1544–1551.
- Fuchs, T., Chowdhury, G., Barnes, C. L. & Gates, K. S. (2001). J. Org. Chem.66, 107–114. [DOI] [PubMed]
- Ley, K. & Seng, F. (1975). Synthesis, 415, 415–422.
- Monge, A., Palop, J. A., Lopez de Cerain, A., Senador, V., Martinez-Crespo, F. J., Sainz, Y., Narro, S., Garcia, E., Miguel, C., Gonzalez, M., Hamilton, E., Barker, A. J., Clarke, E. D. & Greenhow, D. T. (1995). J. Med. Chem.38, 1786–1792. [DOI] [PubMed]
- Mustaphi, N. E., Ferfra, S., Essassi, E. M. & Pierrot, M. (2001). Acta Cryst. E57, o176–o177.
- Oxtoby, N. S., Blake, A. J., Champness, N. R. & Wilson, C. (2005). Chem. Eur. J.11, 4643–4654. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siemens (1991). P3/PC Data Collection Software Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Zarranz, B., Jaso, A., Aldana, I. & Monge, A. (2004). Bioorg. Med. Chem.12, 3711–3721. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808003784/hk2425sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808003784/hk2425Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report