

Abstract
Herpes simplex virus type 1 (HSV-1) infected cell protein 0 (ICP0) is a multifunctional protein that functions as a promiscuous transactivator and promotes the degradation of multiple cellular proteins. In vitro studies indicated that it encodes two physically separated functional E3 ubiquitin ligase domains. One, designated herpesvirus ubiquitin ligase 1 (HUL-1), maps to a region encoded by exon 3 and is contained between residues 543 and 680. Deletion of amino acids 621 to 625 abolishes this activity. The second, designated HUL-2, maps to the RING finger domain present in ICP0 encoded by exon 2. Earlier studies have shown that ICP0 stabilizes cyclins D1 and D3, and several lines of investigation led to the hypothesis that this function of ICP0 is the consequence of degradation of the E2 enzyme cdc34, known to be involved in the proteasome-dependent degradation of D-type cyclins. Consistent with this hypothesis, we have previously shown that cdc34 physically interacts with ICP0 at or near aspartate 199 and at amino acids 621 to 625 and that the former site is required for effective ubiquitylation and degradation of cdc34. Furthermore, the ICP0 HUL-1 domain promotes the polyubiquitination of cdc34 in vitro. If the mechanism by which D-type cyclins are salvaged in wild-type-infected cells is dependent on polyubiquitination and consequent destruction of cdc34, than the mutant virus R6701, which was constructed for these studies and lacks ICP0 residues 621 to 625, should destabilize the D cyclins and preclude the degradation of cdc34. We report that ICP0 residues 621 to 625 are essential for degradation of cdc34 in infected cells and for the ICP0-mediated stabilization of D-type cyclins, that a mutation that specifically disrupted the ring finger domain of the HUL-2 site had no effect on the degradation of cdc34 in infected cells, and that deletion of ICP0 residues 621 to 625 decreased the replicative capacity of the virus in growth-arrested but not in dividing cells and resulted in diminished pathogenicity on intracerebral inoculation of mice. We conclude that the ICP0 HUL-1 domain acts in infected cells to degrade cdc34 and that this function requires the interaction of cdc34 with sequences in exons 2 and 3 but does not involve the HUL-2 RING finger E3 domain.
Infected cell protein 0 (ICP0), the major protein product of the herpes simplex virus type 1 (HSV-1) α0 gene, is a promiscuous transactivator shown to enhance the expression of genes introduced into cells by infection or transfection (12, 13, 23, 44, 49). ICP0 is critical for viral replication in cultured cells infected at low multiplicity, but is not essential in cells infected at high multiplicity (53, 58). The ICP0 protein is 775 amino acids in length and is translated from a spliced mRNA containing three exons encoding 19, 222, and 534 codons (reviewed in reference 50). While the amino acid sequence is not particularly informative, analysis of the arrangement of cysteine and histidine residues in ICP0 exon 2 revealed that in encodes a (RING) finger domain (amino acids 116 to 156) (18, 21). All available evidence suggests ICP0 is a multifunctional protein and that its role in viral infection reflects the sum of its multiple and diverse functions (50). An extensive literature has documented several important properties of ICP0 which are described in the paragraphs below.
ICP0 has been shown to interact with a number of cellular proteins, including the transcription factor BMAL-1 (31), translation elongation factor 1δ (EF-1δ) (32), cyclin D3 (30, 60, 62), a protein designated as p60 (7), and the ubiquitin-specific protease 7 (USP7) (16, 17, 41, 42). Early in infection, ICP0 localizes to ND10 nuclear structures containing the promyelocytic leukemia protein (PML) and effects their disruption (15, 38, 39). Later in infection, ICP0 is translocated from the nucleus to the cytoplasm (32, 37, 60).
In recent years, several lines of evidence suggested that ICP0 is involved in the degradation of cellular proteins. In infected cells, ICP0 colocalizes with conjugated ubiquitin (15, 46) and mediates the degradation of CENP-C (20), CENP-A (36), Sp100 (9, 45), the catalytic subunit of DNA dependent protein kinase (DNA-PKCS) (33, 47), PML, especially sumoylated isoforms (19), and other hitherto unidentified sumoylated proteins (19) in a RING finger domain-dependent manner. In the course of our studies, several lines evidence suggested that ICP0 acts as a ubiquitin ligase and is involved in the degradation of the E2 ubiquitin-conjugating enzyme cdc34 (UbcH3).
cdc34 came to be suspected as a target for degradation by ICP0 for the following reasons. (i) It is the major E3 enzyme interacting with the Skp1-cdc34-F-box (SCF) E2 ubiquitin ligase complex (28), and SCF malfunction has been linked to the accumulation of cyclin D1 in certain tumors (22, 51, 52). In HSV-1-infected cells, both cyclin D1 and cyclin D3 are stabilized even though ICP0 interacts only with cyclin D3 (30, 60, 62). (ii) cdc34 physically interacts with ICP0 at two sites, at or near aspartate 199 (D199) (27) and at or near residues 621 to 625 (26). Neither cyclin D1 nor cyclin D3 is stabilized in cells infected with a mutant virus carrying a point mutation in ICP0 in which aspartate 199 is replaced with alanine (D199A) (60, 62). (iii) ICP0 interacts dynamically with proteasomal subunits (37, 61) (iv) In infected cells, cdc34 is degraded by the 26S proteasome and ubiquitylated isoforms of cdc34 bind to proteasomal subunits in the presence of the proteasome inhibitor MG132 in a manner dependent on ICP0 and specifically aspartate 199 (27, 61).
Encouraged by these observations, this laboratory demonstrated that, in in vitro ubiquitylation reactions, a segment of ICP0 encoded by residues 543 to 680 has E3 ubiquitin ligase activity in conjunction with the E2 enzyme cdc34 but not E2 enzymes UbcH5a, UbcH6, and UbcH7 and promotes the autoubiquitylation of cdc34 (26, 27, 61). Further studies showed that deletion of amino acids 621 to 625 abrogated the E3 activity of this region in vitro (26). We have designated this E3 site encoded by ICP0 exon 3 herpesvirus ubiquitin ligase 1 (HUL-1).
The model emerging from these studies is that cdc34 is polyubiquitinated and degraded in order to preclude the degradation of D-type cyclins by the SCF complex (24). One prediction of this model is that in cells infected with a mutant virus (R6701) lacking ICP0 amino acids 621 to 625, cdc34 would not be degraded but D-type cyclins would be degraded. In this report we show that this is in fact the case. We also demonstrate that growth of R6701 is attenuated in resting cells but not in dividing cells and that R6701 exhibits reduced neurotoxicity in an experimental animal system. These results indicate that amino acids 621 to 625 are indispensable for the function of HUL-1.
We should note that subsequent to the discovery of the HUL-1 it became apparent that ICP0 contains a second ubiquitin ligase site (HUL-2), mapping to the RING finger domain, that functions in conjunction with the E2 ubiquitin ligase enzymes UbcH5a and UbcH6 (5, 27). In a more recent study, it was demonstrated that the functional interaction of ICP0 with UbcH5a is responsible for the proteasome-dependent degradation of PML and Sp100 (25), indicating that RING finger E3 function mediates the degradation of these proteins.
MATERIALS AND METHODS
Cells and viruses.
SK-N-SH, Vero, and HeLa cell lines were obtained from the American Type Culture Collection (Manassas, Va.), and rabbit skin cells were originally obtained from J. McClaren (University of New Mexico, Albuquerque). Telomerase-transformed human diploid foreskin fibroblasts (HFF) (6) were obtained from Thomas Shenk (Princeton University, Princeton, N.J.). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (HFF cells, SK-N-SH cells) or 5% newborn calf serum (rabbit skin cells, Vero cells, HeLa cells). HSV-1 strain F [HSV-1(F)], a limited-passage isolate, is the prototype HSV-1 strain used in this laboratory (11).
Recombinant viruses containing mutations on an HSV-1(F) background, R7910 (Δα0, ICP0 null) (30) and R7914 (ICP0 D199A) (62), have been described previously. Recombinant virus vC116G/C1656A, encoding ICP0 C116G/C156A on an HSV-1 strain 17 [HSV-1(17)] background, was obtained from Saul Silverstein (Columbia University, New York, N.Y.), and its construction and phenotypic properties were described elsewhere (35). All virus stocks used in this study were titered on Vero cells by standard methods with the exception of R7910, which was titered on U20S cells.
Serum starvation and contact inhibition of HFF cells.
HFF cells were arrested in G1 phase by serum starvation as previously described (60), and 50% confluent cultures of HFF cells were rinsed with phosphate-buffered saline (PBS) and incubated at room temperature for 7 days in DMEM supplemented with 0.25% fetal bovine serum. The cultures were incubated at 37°C in the same medium for 1 day prior to infection with HSV. In contact inhibition experiments, HFF cells cultured in DMEM supplemented with 10% fetal bovine serum were allowed to become fully contact inhibited and were maintained at 37°C for 7 additional days as previously described (62).
Plasmids.
pRB5849 encodes a portion of HSV-1(F) ICP0 consisting of codons 543 to 768 that contains a deletion spanning residues 621 to 625 inserted into vector pGEX 4T-1 in frame with glutathione S-transferase (GST) (26), and pRB3710 encodes a SacI-PstI fragment encompassing the entire ICP0 coding sequence in vector pUC18 (40). The 2,557-bp BamHI-SalI ICP0 fragment from pRB3710 was ligated into a BamHI- and SalI-digested pBluescript II KS+ (Stratagene, La Jolla, Calif.) plasmid to yield pRB5919. The AatII-BsiWI fragment of pRB5849 containing the ICP0 Δ621-625 deletion in ICP0 exon 3 was purified and ligated to an AatII- and BsiWI-digested pRB5919 plasmid. The resulting plasmid contained the Δ621-625 deletion in the ICP0 fragment encoded by pRB5919 and was designated pRB5920. pRB5920 was digested with BamHI and SalI, and the resulting ICP0 fragment containing the Δ621-625 deletion was ligated to a BamHI- and SalI-digested pRB3710 plasmid. The resulting plasmid containing the Δ621-625 deletion in the full-length ICP0 coding sequence encoded by pRB3710 was designated pRB5921.
Plasmid pRB5270, designed to restore ICP0 coding sequences within the ICP0 null recombinant virus R7910, carrying the HSV-1(F) BsrGI fragment encoding the ICP0 gene and flanking sequences cloned in the Acc65I site of vector pUC19, was previously described (62). A SnaBI-BsMI fragment of pRB5921 was purified and ligated to a SnaBI- and BsMI-digested pRB5270 plasmid. The resultant plasmid encoding the Δ621-625 deletion in the ICP0 sequence encoded by pRB5270 was designated pRB5922 and was sequenced at the University of Chicago Cancer Research Center DNA sequencing facility (Chicago, Ill.) to confirm that the ICP0 sequence it encoded contained the deletion.
Construction of recombinant virus R6701.
Recombinant virus R6701 was constructed by transfection of rabbit skin cells with 2 μg of pRB5922 with Lipofectamine Plus (Gibco BRL, Rockville, Md.) according to the manufacturer's instructions, followed by superinfection with R7910 at a multiplicity of infection of 0.05, 0.01, and 0.001 after 24 h as described previously (62). After 2 days, superinfected cultures were frozen at −80°C, thawed, sonicated and titered on Vero cells. Large plaques were isolated from dishes containing 20 to 50 plaques and grown on Vero cells. Viral isolates were screened for ICP0 expression by probing Western blots of electrophoretically separated lysates of infected Vero cells with antibody directed against ICP0. After the initial screen, an additional round of plaque purification and screening for ICP0 expression was performed.
HSV-1 infections and preparation of cell lysates.
Cultures of the indicated cell type were exposed to the indicated number of PFU per cell of the appropriate virus in medium 199V (medium 199 supplemented with 1% calf serum) on a rotary shaker at 37°C in all cases except for experiments examining viral growth in serum-starved cells, in which DMEM supplemented with 0.25% fetal bovine serum was used in the inoculum instead of medium 199V. After 1 or 2 h, the inoculum was replaced with fresh growth medium. For experiments involving MG132 treatment, the inoculum was replaced with growth medium containing 10 μM MG132 (BioMol, Plymouth Meeting, Pa.) for the duration of time indicated.
For electrophoretic separation of cell lysates (see below), cells were harvested at the indicated time after infection, washed twice in PBS, pelleted by centrifugation, and solubilized at 4°C in PBS-A* (1% Nonidet P-40, 1% deoxycholate in PBS) in which complete Mini EDTA-free protease inhibitor cocktail tablets (Roche Diagnostics, Mannheim, Germany) had been dissolved according to the manufacturer's instructions. Lysates were then sonicated briefly. The amount of total protein in each lysate was quantified with the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, Calif.) according to the manufacturer's instructions. To prepare samples for electrophoresis, 1 part 4× disruption buffer (6.85% sodium dodecyl sulfate, 24% glycerol, 3.3% β-mercaptoethanol, 233 mM Tris, pH 6.8, 0.008% bromophenol blue) was then added to 3 parts lysate. For viral growth experiments, cultures were frozen at −80°C at 20 h after infection, thawed, sonicated, and titered on Vero cells. Virus yields were determined by standard plaque assays.
Electrophoretic separations, immunoblotting, and antibodies.
Electrophoresis and immunoblotting procedures used in these studies have been described elsewhere (61). Samples were boiled for 5 min, and solubilized proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on bisacrylamide gels, transferred to nitrocellulose sheets, blocked with 5% nonfat milk, and reacted with primary antibody followed by the appropriate secondary antibody conjugated to alkaline phosphatase (Bio-Rad) or horseradish peroxidase (Sigma, St. Louis, Mo.). Immunoblots were developed with enhanced chemiluminescence (ECL) (Amersham Biosciences, Piscataway, N.J.) or 5-bromo-4-chloro-3-indolylphosphate (BCIP)/nitroblue tetrazolium (NBT) (Sigma) accordingly.
Mouse monoclonal antibodies against ICP4 and ICP27 (1) were purchased from the Goodwin Cancer Research institute (Plantation, Fla.) and used at a dilution of 1:500 for immunoblots. Rabbit polyclonal antibody raised against ICP0 exon 2 was used at a dilution of 1:1,000 as described elsewhere (30). Mouse monoclonal antibodies against cdc34 (catalog no. 610250), cyclin D1 (catalog no. 556470), and cyclin D3 (catalog no. 554195) were purchased from BD PharMingen (San Diego, Calif.) and all were used at a dilution of 1:1,000.
Intracranial inoculation of mice with HSV-1.
Five-week-old CBA/J mice (The Jackson Laboratory, Bar Harbor, Maine) were anesthetized with 100 mg of nembutal (Abbott Laboratories, Abbott Park, Ill.). Mice were injected intracranially with 50 μl of virus inoculum diluted in sterile PBS. The virus was allowed to absorb for 5 s before needle retraction. Mice were checked for mortality twice daily from day 2 to day 25. The 50% lethal dose was calculated by the trimmed Spearman-Karber method with the trimmed Spearman-Karber program, version 1.5 (Ecological Monitoring Research Division, Environmental Monitoring Systems Laboratory, U.S. Environmental Protection Agency, Cincinnati, Ohio). All mice were treated in accordance with institutional guidelines for the humane care and treatment of animals.
RESULTS
Preparation of reagents.
In an earlier report it was shown that in in vitro studies, ICP0 lacking residues 621 to 625 destroyed the HUL-1 E3 activity and disrupted the interaction of ICP0 exon 3 with cdc34 in vitro (26). To determine the phenotype of a virus mutant carrying this mutation in both copies of ICP0, we constructed R6701, a recombinant virus carrying the Δ621-625 deletion in ICP0 by rescuing growth of ICP0 null virus in rabbit skin cells with ICP0 Δ621-625 as previously described (62).
As described in Materials and Methods, the recombinant virus R6701 was isolated from viral progeny obtained after exposure to R7910 (Δα0 mutant) at low multiplicities of infection of rabbit skin cells previously transfected with plasmid pRB5922 encoding ICP0 Δ621-625 and flanking regions. The rationale behind this approach was as follows. Growth of Δα0 virus in rabbit skin cells is severely impaired at low multiplicities of infection. As ICP0 Δ621-625 lacks only 5 of the 775 amino acids in wild-type ICP0, it could be expected that ICP0 Δ621-625 would encode many of the functions of ICP0, and thus, a virus expressing ICP0 Δ621-625 would have a significant growth advantage over an ICP0 null virus. Thus, in the population of progeny produced by the superinfected cells, homologous recombination events between the regions flanking the ICP0 locus in ICP0 null viral DNA and the pRB5922 plasmid resulting in incorporation of ICP0 Δ621-625 into the viral genome are selected for.
In electrophoretically separated lysates of infected cells, ICP0 protein expressed by R6701 is readily detectable and migrates slightly faster than that expressed by wild-type virus (Fig. 1, compare lanes 2 and 3). This observation is consistent with the presence of the small deletion in ICP0 encoded by R6701. In addition, both ICP0 and ICP0 Δ621-625 are present in infected cell lysates in comparable amounts (Fig. 1, lanes 2 and 3), suggesting that deletion of residues 621 to 625 does not appreciably affect ICP0 synthesis or stability.
FIG. 1.
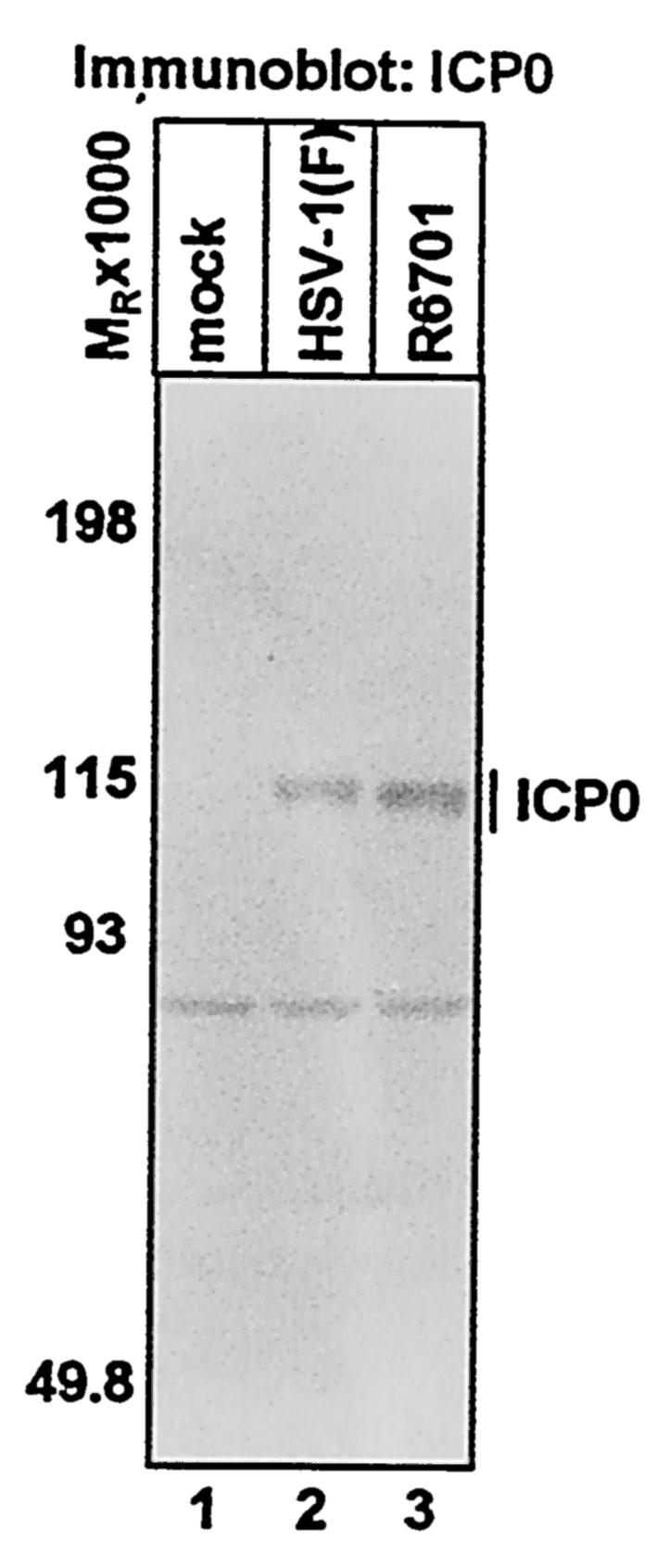
Accumulation of wild-type and mutant ICP0 (Δ621-625) in infected cells. SK-N-SH cells were either mock infected (lane 1) or exposed to 5 PFU of HSV-1(F) (lane 2) or R6701 (lane 3) per cell for 1 h and harvested at 5 h after infection. Cells were then solubilized, and lysates containing 80 μg of total protein were electrophoretically separated. Proteins were transferred to nitrocellulose sheets and probed with a rabbit polyclonal antibody against ICP0.
ICP0-dependent degradation of cdc34 in vivo requires ICP0 residues 621 to 625 and is mediated by the HUL-1 E3 ligase encoded by ICP0 exon 3.
To test the hypothesis that ICP0 exon 3 E3 activity promotes the degradation of cdc34 in vivo, we examined cdc34 protein levels at 5 h after infection in SK-N-SH cells that were mock infected or exposed to 10 PFU of HSV-1(F) or the recombinant virus R6701 (ICP0 Δ621-625) per cell by reacting electrophoretically separated proteins with a monoclonal antibody against cdc34. Decreased amounts of cdc34 accumulate in cells infected with HSV-1(F) compared to mock-infected cells (Fig. 2A, compare lanes 1 and 2). Treatment of infected cells with MG132 results in significantly more cdc34 accumulation than occurs in untreated infected cells (Fig. 2A, compare lanes 2 and 3) indicating that cdc34 is degraded in a proteasome-dependent manner in infected cells. However, no reduction in cdc34 accumulation compared to mock-infected cells or increase in cdc34 levels in cells treated with MG132 is observed in cells infected with R6701 (Fig. 2A, lanes 4 and 5), indicating that this virus is defective for the ability to promote degradation of cdc34.
FIG. 2.

Degradation of the E2 enzyme cdc34 in HSV-1-infected cells requires ICP0 residues 621 to 625 and is independent of the ring finger domain encoded by exon 2. SK-N-SH cells were either mock infected (lane 1) or exposed to 10 PFU of HSV-1(F) (lanes 2 and 3), R6701 (lanes 4 and 5), or vC116G/C156A (lanes 6 and 7) per cell for 1 h. The virus inoculum was removed, and cells were incubated in growth medium or growth medium containing 10 μM MG132 as indicated for 4 h before harvest at 5 h after infection. Cells were solubilized, and lysates containing 80 μg of total protein were subjected to electrophoresis in denaturing polyacrylamide gels. Electrophoretically separated proteins were transferred to nitrocellulose sheets and reacted with mouse monoclonal antibody against cdc34.
As noted in the introduction, a second E3 ligase activity was mapped to the RING finger domain encoded by exon 2. It was therefore of interest to determine whether ICP0 RING finger E3 activity played a role in ICP0-mediated degradation of cdc34. To address this issue, we examined cdc34 protein levels in cells exposed to 10 PFU per cell of vC116G/C156A, a recombinant virus encoding C116G and C156A point mutations in ICP0, both of which destroy Zn2+ binding sites in the ICP0 RING finger, ablating its E3 ligase activity (35; R. Hagglund, S. Sliverstein, and B. Roizman, unpublished results). cdc34 is effectively degraded in a proteasome-dependent manner in cells infected with the vC116G/C156A mutant (Fig. 2A, lanes 6 and 7). We conclude from this experiment that an intact RING finger domain and ICP0 RING finger-dependent E3 ligase activity are not required for ICP0-mediated degradation of cdc34, consistent with the hypothesis that the discrete E3 function encoded by ICP0 exon 3 mediates cdc34 destruction. As the viral α proteins ICP27 and ICP4 accumulated in comparable amounts in HSV-1(F)-, R6701-, and vC116G/C156A-infected cells (Fig. 2B and data not shown), the observed differences in cdc34 accumulation cannot be attributed to exposure of cells to different amounts of HSV-1. We conclude from these experiments that ICP0 residues 621 to 625 are required for ICP0 to promote the degradation of cdc34 indicating that it is mediated by ICP0 exon 3 HUL-1 E3 activity.
Deletion of residues 621 to 625 disrupts the ability of ICP0 to promote the stabilization of cyclin D1 and cyclin D3.
Given the result that cdc34 is stabilized in cells infected with a virus mutant in which ICP0 lacks residues 621 to 625 (Fig. 2), the model discussed in the introduction predicts that cyclins D1 and D3 would be degraded in cells infected with this mutant, that is, exactly the opposite of what happens in wild-type virus infected cells. The accumulation of D-type cyclins is very much cell type dependent (3 and data not shown). For instance, ample amounts of cyclin D3 accumulate in uninfected Vero cells (Fig. 3A, lane 1), while little if any cyclin D1 is detectable in this cell line (data not shown). Thus, to test the hypothesis described above, two series of experiments were performed.
FIG. 3.

ICP0 residues 621 to 625 are necessary for ICP0-dependent stabilization of D-type cyclins in infected cells. (A) Vero cells were either mock infected (lane 1) or exposed to 0.5 PFU of HSV-1(F) (lanes 2, 6, and 10), R6701 (ICP0 Δ621-625) (lanes 3, 7, and 11), R7910 (Δα0) (lanes 4, 8, and 12), or R7914 (ICP0 D199A) (lanes 5, 9, and 13) per cell. The cells were harvested at 6 (lanes 2 to 5), 10 (lanes 6 to 9), or 12 (lanes 10 to 13) h after infection and solubilized. Lysates containing 75 μg of total protein were subjected to SDS-PAGE. Electrophoretically separated proteins were transferred to nitrocellulose sheets and reacted with mouse monoclonal antibodies against cyclin D1, cyclin D3, or ICP4. (B) Serum-starved HFF were either mock infected (lane 1) or exposed to 10 PFU of HSV-1(F) (lanes 2 to 9) or R6701 (lanes 10 to 17) per cell. The cells were harvested at the times indicated, lysates containing 75 μg of total proteins were harvested, and separated proteins were reacted with anti-cyclin D1 mouse monoclonal antibody as described above.
In the first, replicate cultures of Vero cells were either mock infected or exposed to 0.5 PFU of HSV-1(F), R6701 (ICP0 Δ621-625), R7910 (ICP0 null), or R7914 (ICP0 D199A). In earlier studies, this laboratory has shown that D-type cyclins are not stabilized in cells infected with recombinant viruses R7910 (30, 62) and R7914 (60, 62). After cells were harvested and solubilized at 6, 10, and 12 h after infection, cell lysates were subjected to electrophoresis in denaturing gels and reacted with monoclonal antibody against cyclin D3.
The results (Fig. 3A) were as follows. At 6 h after infection, comparable amounts of cyclin D3 were observed in cells infected with HSV-1(F), R6701, R7910, and R7914 (Fig. 3A, lanes 2 to 5). In cells infected with wild-type virus, a robust amount of cyclin D3 was present at 10 h after infection, and a significant amount of cyclin D3 was still detectable at 12 h after infection (Fig. 3A, lanes 6 and 10). In contrast, the amount of cyclin D3 present in cells infected with R6701 was severely decreased compared to that observed in cells infected with wild-type virus (Fig. 3A, compare lanes 6 and 7). Moreover, cyclin D3 could not be detected in R6701-infected cells at 12 h after infection (Fig. 3A, lane 11), indicating that deletion in ICP0 carried by R6701 also precluded the ICP0-dependent stabilization of cyclin D3 in Vero cells. Consistent with previous results (30, 62), these results indicate that R7910 and R7914 are defective for stabilization of cyclin D3 (Fig. 3A, lanes 8, 9, 12, and 13). Since ICP4 accumulated to comparable amounts in all samples infected with HSV-1 (Fig. 3A), differences in cyclin D accumulation cannot be attributed to exposure of cells to different amounts of HSV-1 virus.
In the second series of experiments, replicate cultures of serum-starved quiescent HFF cells were either mock infected or exposed to 10 PFU of HSV-1(F) or R6701 per cell, harvested at 1, 3, 4, 5, 6, 8, 10, or 12 h after infection, and lysed. Cell lysates were separated by electrophoresis in denaturing gels, and the separated proteins were reacted with monoclonal antibodies against cyclin D1 and cyclin D3. In contrast to what was observed in Vero cells (Fig. 3A), both cyclin D1 and cyclin D3 were present in uninfected HFF lysates (Fig. 3B and data not shown). Compared to cells infected with wild-type virus, cyclin D1 levels were significantly decreased by 4 h after infection (Fig. 3B, compare lanes 4 and 12), and cyclin D1 was undetectable at 6 h after infection in R6701-infected cells (Fig. 3B, lane 14), while it was readily detected at this time point in cells infected with HSV-1(F) (Fig. 3B, lane 6). These results indicate that the kinetics of cyclin D1 accumulation during HSV-1 infection in HFF differ from those in Vero cells. In addition, disappearance of cyclin D3 was also accelerated in cells infected with R6701 compared to those infected with wild-type virus (data not shown). ICP4 accumulated to comparable amounts with identical kinetics in cells infected with either HSV-1(F) or R6701 (Fig. 3B), indicating that differences in cyclin D1 levels in cells infected with these viruses cannot be attributed to exposure of cells to different amounts of virus or a defect in α protein accumulation in R6701. We conclude from these results that ICP0 residues 621 to 625 are necessary for ICP0-mediated stabilization of cyclin D1 and cyclin D3 during HSV-1 infection.
Deletion of ICP0 residues 621 to 625 results in impaired replication of HSV-1 in resting cells but not in actively dividing cells.
These experiments were designed to characterize the replicative properties of recombinant virus R6701 (ICP0 Δ621-625) in cultured cells. In the first series of experiments, we examined the replication of R6701 in rabbit skin cells and two primate cell lines (Vero and HeLa cells). To account for the observation that the replication of ICP0 null viruses is multiplicity dependent (53, 58), cells were exposed to either 0.05 PFU or 5 PFU of HSV-1(F) or R6701 per cell and harvested 20 h after infection. As illustrated in Table 1, there was no significant difference in the yield of HSV-1(F) or R6701 from rabbit skin, Vero, or HeLa cells exposed to the low or high ratios of virus to cells.
TABLE 1.
Replication of HSV-1(F) and ICP0 Δ621-625 virus in primate and rabbit cell linesa
| Cell line | Infection (PFU/cell) | Yield (PFU, 107)
|
|
|---|---|---|---|
| HSV-1(F) | R6701 | ||
| RSC | 0.05 | 57 | 20 |
| 5 | 200 | 90 | |
| HeLa | 0.05 | 3.1 | 1.5 |
| 5 | 150 | 450 | |
| Vero | 0.05 | 18 | 21 |
| 5 | 190 | 370 | |
Cells were grown in 25-cm2 flasks, exposed to either 0.05 or 5 PFU of HSV-1 per cell as indicated, and harvested at 20 h after infection.
The second series of experiments took into account the observation that some HSV-1 mutants, like R7914 (ICP0 D199A), replicate better in actively dividing cells than in growth-arrested, quiescent primary fibroblasts (62). In this series of experiments, replicate subconfluent cultures of dividing HFF cells or confluent, serum-starved cultures or confluent, contact-inhibited cultures as described in Materials and Methods were exposed to 0.05 PFU of the wild type or R6701 mutant per cell. As shown in Table 2, infection with R6701 yielded >10-fold less virus in contact-inhibited or serum-starved HFF fibroblasts than infection with wild-type virus. In contrast, there were no significant difference in the yields of HSV-1(F) or R6701 from actively dividing HFF cells (Table 2). These results indicate that the attenuation of R6701 observed in serum-starved and contact-inhibited HFF cells was due to their resting state and not explained by a general growth defect in fibroblasts.
TABLE 2.
Replication of wild-type virus and ICP0 Δ621-625 in actively dividing and resting pHF fibroblastsa
| pHF cells | Yield (PFU, 105)
|
|
|---|---|---|
| HSV-1(F) | R6701 | |
| Actively dividing | 1,500 | 1,300 |
| Serum starved | 70 | 5.0 |
| Contact inhibited | 800 | 29 |
Diploid HFF fibroblasts grown in 25-cm2 flasks were infected with HSV-1 while actively divding before reaching confluence or either allowed to become fully confluent (contact inhibited) and maintained in the same medium for 7 days or serum starved in medium containing 0.25% fetal calf serum for 5 days prior to infection. Cells were exposed to 0.05 PFU of HSV-1 per cell in the same medium in which they were maintained prior to infection and harvested at 20 h after infection.
Function of ICP0 residues 621 to 625 contributes to the virulence of HSV-1.
In this series of experiments, we examined the effect of the deletion of these residues on the capacity of the virus to replicate in and destroy the central nervous system (CNS) (neurotoxicity). As destruction of the CNS and death of the animal are a direct consequence of virus multiplication in the CNS (50), the readout for CNS pathology used in these experiments is death of the animal. In these experiments, 5-week-old CBA/j mice were inoculated intracerebrally with serial 10-fold dilutions of HSV-1(F), R6701 (ICP0 Δ621-625), or R7910 (Δα0) in groups of six mice per dilution. The 50% lethal doses, calculated as described in Materials and Methods, were 1.0 × 102 for HSV-1(F), 4.6 × 103 for R6701, and >106 for the Δα0 mutant R7910. These results indicate a significant decrease in neurotoxicity of greater than 45-fold of the mutant virus expressing ICP0 Δ621-625 compared with wild-type virus. Furthermore, the defect in neurotoxicity exhibited by the ICP0 null virus is much more pronounced, inasmuch as this virus displayed at least a 10,000-fold decrease in neurotoxicity compared to wild-type virus.
In a parallel series of experiments, we examined the length of time required for HSV-1(F) and R6701 to induce CNS pathology in neurotoxicity studies in an experimental animal system. The speed at which a given virus is able to induce CNS pathology after intracranial injection is an important aspect of virulence because it measures the ability of the virus to replicate effectively in the CNS, leading to its destruction. To these ends, intracerebral inoculation of 5-week-old CBA/j mice with 100 PFU of HSV-1(F) or R6701 per mouse in groups of nine mice per virus was performed. Among animals that succumbed to HSV-1 infection, the mean time to death of those inoculated with wild-type virus was 6.5 days, while it was 16.5 days for animals inoculated with R6701. These results are consistent with the hypothesis that the deletion of ICP0 amino acids 621 to 625 reduces the capacity of the virus to multiply in and destroy the CNS of mice.
DISCUSSION
The series of studies leading to this report stemmed from the observation that cyclin D3 interacts with ICP0 in a yeast two-hybrid system and in vitro and that, in cells infected with wild-type virus, the cyclin D3 is stabilized, whereas in cells infected with the Δα0 mutant, cyclin D3 disappears very rapidly (30, 62). A single amino aid substitution (D199A) in ICP0 exon 2 abolished both binding and stabilization of cyclin D3 by ICP0 (60, 62). Cyclin D1 did not interact with ICP0 either in the yeast-two-hybrid system or in vitro (60). Nevertheless, like cyclin D3, cyclin D1 is stabilized in wild-type virus-infected cells but not in cells infected with a recombinant virus expressing ICP0 D199A (60). These observations raised two questions, i.e., the function of cyclin D3 in infected cells and the mechanism by which cyclin D3 and D1 were stabilized even though only cyclin D3 interacted with ICP0 in the systems in which the interactions were tested.
With respect to the first question, the known function of D-type cyclins is to mediate the transition from G1 to S phase. Extensive studies have shown that HSV-1 does not drive the cell into S phase (2, 10, 57). Other studies, however, demonstrated that ICP0 is transported to the cytoplasm after the onset of viral DNA synthesis (32, 37, 60) and that the D199A substitution in ICP0 blocked translocation of the protein, whereas overexpression of cyclin D3 accelerated the translocation of ICP0 to the cytoplasm (57). cdk4, the D-type cyclin-dependent kinase, is activated in HSV-1-infected cells in a manner dependent on ICP0 aspartate 199 (60). Thus, it is likely that stabilization of D-type cyclins alters the cellular environment, making it more conducive to viral growth by allowing the virus to use cdk4 activity to benefit viral replication.
Among herpesviruses, HSV-1 is not unique in that D-type cyclins play a role in its life cycle. Epstein-Barr virus encodes proteins that induces expression of D-type cyclins (56), and human herpesvirus 8 and herpesvirus saimiri encode functional homologs of D-type cyclins (8, 29, 34, 43).
The route to answering the second question, the mechanism by which both cyclin D1 and cyclin D3 were stabilized, was more tortuous. One clue emerged from the observation that both cyclins were stabilized even though ICP0 interacted with cyclin D3 only (60). The second clue emerged from the observation that ICP0 interacted dynamically with proteasome components and was also bound to proteasomes in the presence of MG132, a proteasome inhibitor (37, 61). This behavior suggested that ICP0 may be a component of the ubiquitin-proteasome pathway, and the presence of a RING finger domain in exon 2 suggested that it may act as an E3 ubiquitin ligase. If it were a ubiquitin ligase, one mechanism by which ICP0 could block the degradation of both cyclins would be to target a component of the ubiquitin ligase system responsible for the degradation of D cyclins for destruction. Defects in ubiquitylation machinery components result in cyclin D1 stabilization in certain human tumors (22, 51, 52). cdc34 is the major ubiquitin-conjugating enzyme linked to the degradation of D-type cyclins in conjunction with SCFSkp2 E3 ubiquitin ligase (Fig. 4A) (22, 28, 51, 52, 63). In in vitro assays, however, it was exon 3 and not exon 2 that polyubiquinated cdc34 in an ATP-dependent fashion (26, 27, 61). Further studies established that exon 3 binds cdc34 and that amino acids 621 to 625 are essential for the polyubiquitination and for binding of cdc34 by ICP0 (26, 61). Moreover, in wild-type virus-infected cells, cdc34 is degraded, while it is not degraded in cells infected with a recombinant virus expressing ICP0 Δ621-625 (Fig. 2). In the proposed model, degradation of cdc34 mediated by ICP0 (Fig. 4B) would preclude the degradation of D-type cyclins (Fig. 4C).
FIG. 4.

Schematic representation of the degradation of cdc34 and stabilization of D-type cyclins effected by ICP0. (A) The SCF E3 complex promotes the ubiquitylation and subsequent degradation of D-type cyclins (adapted from reference 59). D-type cyclins are recruited to the complex by the F-box protein Skp2 via its leucine-rich repeat (LRR) domain (22, 51, 63). Other F-box proteins recruit different substrates (reviewed in reference 25). The F-box protein interacts with SCF components Skp1 and the RING finger protein Rbx1. The cullin (cdc53/CUL1) forms the backbone of the complex and binds Skp1, Rbx1, and the E2 cdc34, which also interacts with the Rbx1 RING finger. The RING finger allosterically activates the E2, cdc34, leading to transfer of ubiquitin to cyclin D1. Degradation of cyclin D3 occurs by the same mechanism (51). (B) Degradation of cdc34 mediated by ICP0 (adapted from reference 26). The HUL-1 domain of ICP0 has E3 activity in conjunction with cdc34 and interaction of the HUL-1 domain with cdc34 is required for E3 activity as evidenced by the finding that residues 621 to 625 are required for both functions. cdc34 also interacts with ICP0 in the region encoded by exon 2 such that aspartate 199 is required for optimal interaction. The model proposes that the interaction site encoded by ICP0 exon 2 tethers cdc34 to the HUL-1 domain. In this way, E2 activity can be efficiently stimulated by HUL-1 E3 activity via the interaction between cdc34 and the HUL-1 domain leading to the efficient autoubiquitylation of cdc34. (C) Model for D-type cyclin stabilization resulting from ICP0-dependent degradation of cdc34. cdc34 is the major E2 enzyme which interacts with the SCF E3 complex (28). Interaction of an E3 with its cognate E2 is seminal to its ability to promote ubiquitylation of substrates. In this model, degradation of cdc34 renders it unavailable to interact with SCF and promote the ubiquitylation of D-type cyclins. Thus, the model predicts that stabilization of D-type cyclins is a consequence of the failure of the ubiquitylation machinery to target them for proteasome-dependent degradation due to the absence of cdc34.
Polyubiquitination of a ubiquitin-conjugating enzyme by the interacting E3 ligase has been reported both in vitro and in vivo (4, 24, 55). However, to establish that the E3 activity in infected cells is responsible for the degradation of cdc34 and, in consequence, the stabilization of cyclins D3 and D1, we carried out the experiments described in this report. The results show that in cells infected with the mutant lacking residues 621 to 625, cdc34 is stable, whereas cyclins D3 and D1 disappear more rapidly (Fig. 2 and 3). An interesting observation made in this study is that the patterns of accumulations of cyclin D1 in HFF cells and Vero cells are different.
Several points related to the studies described in this report should be noted.
(i) ICP0 is the only protein described to date to exhibit two E3 ligase activities. The first, designated HUL-1, is encoded by exon 3 (26, 27, 61). Its only target found to date is cdc34, and as noted above, one consequence of the degradation of this protein is to preclude the degradation of cyclin D3. The second site, designated HUL-2, maps to the RING finger domain in ICP0 exon 2 (5, 27) and mediates the degradation of several cellular proteins (see the introduction). While ICP0 exon 3 E3 activity might act on additional substrates, the degradation of cdc34, its cognate E2 mediated by ICP0 would render such activity inefficient. Thus, the virus might have evolved two discrete E3 activities with different E2 specificities in ICP0 to ensure the efficient degradation other cellular proteins in addition to cdc34. As summarized in Table 3, the results obtained from this laboratory and that of Roger Everett indicate that the functional interactions of E2 ubiquitin-conjugating enzymes with HUL-1 and HUL-2 E3 sites is highly specific (26, 27, 61). Whereas the HUL-1 domain has E3 activity in conjunction with cdc34 (26, 27, 61), the HUL-2 site functions in conjunction with UbcH5a and UbcH6 (5, 27).
TABLE 3.
E2 specificity profiles of ICP0 exon 3 HUL-1 and ICP0 exon 2 RING finger E3 ubiquitin ligase domainsa
| E2 enzyme | E3 activity
|
|
|---|---|---|
| ICP0 exon 3 (HUL-1) | ICP0 exon 2 (RING) | |
| UbcH1 | − | ND |
| UbcH2 | − | −b |
| cdc34 | +b | −b |
| UbcH5a | −b | +b |
| UbcH5b | − | ND |
| UbcH5c | − | ND |
| UbcH6 | −b | +b |
| UbcH7 | −b | −b |
| UbcH9 | − | − |
| UbcH10 | − | −b |
| UbcH13 | ND | −b |
| UbcH16 | ND | −b |
| Ubc18 | ND | −b |
Substrate-independent in vitro ubiquitylation reactions containing recombinant rabbit E1 (Uba1), ATP, creatine phosphate, creatine phosphokinase, and E2 ubiquitin-conjugating enzymes UbcH1, UbcH2, UbcH5b, UbcH5c, UbcH9, UbcH10, and cdc34 (control) as indicated were performed as previously described (61). E3 activity of the ICP0 exon 3 (HUL-1) and ICP0 exon 2 (HUL-2) E3 ubiqutin ligase domains in conjunction with a given E2 was indicated by the ability of either GST-exon 3 (GST fused in frame with ICP0 residues 543 to 768) or GST-exon 2 (GST fused in frame with ICP0 residues 20 to 241) to promote ubiguitinated-protein ligation above basal levels observed in the absence of E3 activity in substrate-independent ubiquitylation reactions containing that E2. +, ICP0 E3 domain has E3 function in reactions containing a given E2 enzyme; −, ICP0 E3 domain does not have E3 function in conjunction with a given E2 enzyme; ND, not determined.
(ii) cdc34 interacts with ICP0 at two sites, at or near the D199 reside encoded by exon 2 (27, 61) and at or near residues 621 to 625 encoded by exon 3 (26). Whereas deletion of these residues abolish binding at the exon 3 site, the D199A substitution reduces but does not eliminate binding of ICP0 to cdc34 at the site encoded by exon 2. A possible requirement for the D199 site in the polyubiquitination of cdc34 emerged from the observation that the interaction of ubiquitylated cdc34 with proteasomes was ablated in cells infected with the mutant carrying the D199A substitution (61). The sequences at or near D199 may serve as an anchoring site for cdc34 as disruption of this site impairs the polyubiquitination and degradation of the protein (27, 61). Since this or another site at or near D199 also binds cyclin D3 (60, 62), the possibility exists that both proteins compete for a specific site and that polyubiquitination of cdc34 occurs when cyclin D3 is in short supply. In the studies shown in this report, however, disruption of the RING finger has no effect on degradation of cdc34 mediated by exon 3 (Fig. 2).
(iii) All available evidence suggests that interaction of ICP0 with cyclin D3 promotes its translocation to ND10 structures (30, 60). Thus, the interaction of ICP0 with cyclin D3 might be functioning to bring cdk4 activity into ND10 structures to phosphorylate ND10 components. Cyclin D3 might also function to shuttle ICP0 to the nuclear pore or out of the nucleus after ND10 disruption or localization of cyclin D3 to ND10 structures might be a prerequisite for targeting of ICP0 for nuclear export by another mechanism. The role of cyclin D1 in viral infection remains unknown. It is possible that cyclin D1 is an “innocent bystander” inadvertently stabilized as a consequence of ICP0-mediated cdc34 degradation, while cyclin D3 via its interaction with ICP0 functions to promote viral infection.
(iv) Earlier studies have shown that the D199A substitution in ICP0 had no effect on the growth of mutant in dividing cells but yielded >10-fold fewer progeny in contact-inhibited or serum-starved human fibroblast cultures but was not significantly less neurotoxic following intracerebral inoculation of mice (62). In the present study, similar results were obtained in cultured cells with the R6701 mutant lacking ICP0 residues 621 to 625. These comparable phenotypes further support the contentions that ICP0 mediates D-type cyclin stabilization by promoting cdc34 degradation and that degradation of cdc34 is a major function of the ICP0 HUL-1 domain E3 ligase. However, the defect in virulence caused by deletion of ICP0 residues 621 to 625 is more severe than that of the D199A point mutation. An obvious explanation for this observation is that the ICP0 HUL-1 domain promotes the degradation of unidentified substrates mediated by cdc34 inefficiently or by an unknown E2 enzyme. Another explanation involves the fact that the region encoding ICP0 residues 621 to 625 is also required for USP7 binding (16). Thus, the loss of USP7 binding on top of the lack of cdc34 degradation could contribute to the defect in virulence caused be deletion of ICP0 residues 621 to 625. The effect of interfering with the functions of the HUL-1 E3 ligase is far smaller than that observed in cells infected with Δα0 mutants. This observation is consistent with the accumulating evidence that ICP0 is a multifunctional protein and that its phenotype in experimental animal systems reflects the sum of all its functions (50).
(v) It has been reported elsewhere that a double point mutation at arginine 623 and lysine 624 results in a modest (less than 10-fold) defect in viral replication in baby hamster kidney cells (16) which are dividing transformed cells. Like other mutant viruses, such ICP22 null and UL13 null recombinant viruses are defective for growth in rodent cell lines but not primate cell lines (48, 54). Cultured rodent cells are likely a more stringent environment for viral replication than primate cells, in which fewer viral functions are required for optimal replication. Thus, these data are consistent with the data presented in the present study showing that deletion of ICP0 residues 621 to 625 does not significantly affect viral replication in dividing primate and rabbit cells.
ICP0 HUL-1 E3 activity is the latest of the numerous functions encoded by HSV-1 which is dispensable for replication in cultured cells but critical for optimal virulence (50). Knowledge of such functions is seminal for our understanding of the mechanism by which HSV-1 takes total control of the cell to achieve the goal of efficient replication.
Acknowledgments
We thank Charles Van Sant and Guoying Zhou for invaluable advice and Lindsay Smith for expert technical assistance. We also thank Saul Silverstein for the generous gift of the v116G/C156A recombinant virus.
These studies were aided by grants from the National Cancer Institute (CA87661, CA83939, CA71933, CA78766, and CA88860) of the U.S. Public Health Service. While these studies were conducted, R.H. was a Howard Hughes Medical Institute Predoctoral Fellow.
REFERENCES
- 1.Ackermann, M., D. K. Braun, L. Pereira, and B. Roizman. 1984. Characterization of herpes simplex virus 1 alpha proteins 0, 4, and 27 with monoclonal antibodies. J. Virol. 52:108-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Advani, S. J., R. R. Weichselbaum, and B. Roizman. 2000. E2F proteins are posttranslationally modified concomitantly with a reduction in nuclear binding activity in cells infected with herpes simplex virus 1. J. Virol. 74:7842-7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin, H. M., T. J. McDonnell, L. J. Medeiros, G. Z. Rassidakis, V. Leventaki, S. L. O'Connor, M. J. Keating, and R. Lai. 2003. Characterization of 4 mantle cell lymphoma cell lines. Arch. Pathol. Lab. Med. 127:424-431. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee, A., L. Gregori, Y. Xu, and V. Chau. 1993. The bacterially expressed yeast CDC34 gene product can undergo autoubiquitination to form a multiubiquitin chain-linked protein. J. Biol. Chem. 268:5668-5675. [PubMed] [Google Scholar]
- 5.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bresnahan, W. A., G. E. Hultman, and T. Shenk. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruni, R., B. Fineschi, W. O. Ogle, and B. Roizman. 1999. A novel cellular protein, p60, interacting with both herpes simplex virus 1 regulatory proteins ICP22 and ICP0 is modified in a cell type-specific manner and is recruited to the nucleus after infection. J. Virol. 73:3810-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, Y., P. S. Moore, S. J. Talbot, C. H. Boshoff, T. Zarkowska, D. Godden-Kent, H. Paterson, R. A. Weiss, and S. Mittnacht. 1996. Cyclin encoded by KS herpesvirus. Nature 382:410. [DOI] [PubMed] [Google Scholar]
- 9.Chelbi-Alix, M. K., and H. de Thé. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935-941. [DOI] [PubMed] [Google Scholar]
- 10.Ehmann, G. L., T. I. McLean, and S. L. Bachenheimer. 2000. Herpes simplex virus type 1 infection imposes a G1/S block in asynchronously growing cells and prevents G1 entry in quiescent cells. Virology 267:335-349. [DOI] [PubMed] [Google Scholar]
- 11.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 12.Everett, R. D. 1984. Trans activation of transcription by herpes virus products: requirements for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 3:3135-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett, R. D. 1985. Activation of cellular promoters during herpes virus infection of biochemically transformed cells. EMBO J. 4:1973-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everett, R. D. 2000. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 74:9994-10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett, R. D., M. Meredith, and A. Orr. 1999. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 73:417-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett, R. D., M. Meredith, A. Orr, A. Cross, M. Kathoria, and J. Parkinson. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:1519-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everett, R. D., P. Barlow, A. Milner, B. Luisi, A. Orr, G. Hope, and D. Lyon. 1993. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. J. Mol. Biol. 234:1038-1047. [DOI] [PubMed] [Google Scholar]
- 19.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett, R. D., W. C. Earnshaw, J. Findlay, and P. Lomonte. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18:1526-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freemont, P. S. 1993. The RING finger. A novel protein sequence related to the zinc finger. Ann. N. Y. Acad. Sci. USA 684:174-192. [DOI] [PubMed] [Google Scholar]
- 22.Ganiatsas, S., R. Dow, A. Thompson, B. Schulman, and D. Germain. 2001. A splice variant of Skp2 is retained in the cytoplasm and fails to direct cyclin D1 ubiquitination in the uterine cancer cell line SK-UT. Oncogene 20:3641-3650. [DOI] [PubMed] [Google Scholar]
- 23.Gelman, I. H., and S. Silverstein. 1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA 82:5265-5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goebl, M. G., L. Goetsch, and B. Byers. 1994. The Ubc3 (cdc34) ubiquitin-conjugating enzyme is ubiquitinated and phosphorylated in vivo. Mol. Cell. Biol. 14:3022-3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu, H., and B. Roizman. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simpex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 100:8963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagglund, R., and B. Roizman. 2002. Characterization of the novel E3 ubiquitin ligase encoded in exon 3 of herpes simplex virus-1-infected cell protein 0. Proc. Natl. Acad. Sci. USA 99:7889-7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagglund, R., C. Van Sant, P. Lopez, and B. Roizman. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 99:631-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson, P. K., A. G. Eldridge, E. Freed, L. Furstenthal, J. Y. Hsu, B. K. Kaiser, and J. D. R. Reimann. 2000. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 10:429-439. [DOI] [PubMed] [Google Scholar]
- 29.Jung, J. U., M. Stager, and R. C. Desrosiers. 1994. Virus-encoded cyclin. Mol. Cell. Biol. 14:7235-7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawaguchi, Y., C. Van Sant, and B. Roizman. 1997. Herpes simplex virus 1 α regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawaguchi, Y., M. Tanaka, A. Yokoymama, G. Matsuda, K. Kato, H. Kagawa, K. Hirai, and B. Roizman. 2001. Herpes simplex virus 1 α regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc. Natl. Acad. Sci. USA 98:1877-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaguchi, Y., R. Bruni, and B. Roizman. 1997. Interaction of herpes simplex virus 1 α regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J. Virol. 71:1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lees-Miller, S. P., M. C. Long, M. A. Kilvert, V. Lam, S. A. Rice, and C. A. Spencer. 1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70:7471-7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, M., H. Lee, D. W. Yoon, J. C. Albrecht, B. Fleckenstein, F. Neipel, and J. U. Jung. 1997. Kaposi's sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 71:1985-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lium, E. K., and S. Silverstein. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential α27 gene. J. Virol. 71:8602-8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lomonte, P., K. F. Sullivan, and R. D. Everett. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276:5829-5835. [DOI] [PubMed] [Google Scholar]
- 37.Lopez, P., C. Van Sant, and B. Roizman. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 75:3832-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maul, G. G., H. H. Guldner, and J. G. Spivak. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679-2690. [DOI] [PubMed] [Google Scholar]
- 39.Maul, G. G., and R. D. Everett. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75:1223-1233. [DOI] [PubMed] [Google Scholar]
- 40.McKnight, J. L. C., T. M. Kristie, and B. Roizman. 1987. Binding of the virion protein mediating α gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc. Natl. Acad. Sci. USA 84:7061-7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meredith, M., A. Orr, M. Elliott, and R. Everett. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174-187. [DOI] [PubMed] [Google Scholar]
- 42.Meredith, M., A. Orr, and R. D. Everett. 1994. Herpes simplex virus type 1 immediate-early protein Vmw110 binds strongly and specifically to a 135-kDa cellular protein. Virology 200:457-469. [DOI] [PubMed] [Google Scholar]
- 43.Nicholas, J., K. R. Cameron, and R. W. Honess. 1992. Herpesvirus saimiri encodes homologues of G protein-coupled receptors and cyclins. Nature 355:362-365. [DOI] [PubMed] [Google Scholar]
- 44.O'Hare, P., and G. S. Hayward. 1985. Evidence for a direct role for both the 175, 000- and 110, 000-molecular-weight immediate-early gene proteins of herpes simplex virus in the transactivation of delayed-early promoters. J. Virol. 53:751-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parkinson, J., and R. D. Everett. 2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 74:10006-10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parkinson, J., and R. D. Everett. 2001. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 induce the formation of colocalizing, conjugated ubiquitin. J. Virol. 75:5357-5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkinson, J., S. P. Lees-Miller, and R. D. Everett. 1999. Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73:650-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Purves, F. C., W. O. Ogle, and B. Roizman. 1993. Processing of the herpes simplex virus regulatory protein α22 mediated by the UL13 protein kinase determines the accumulation of a subset of α and γ mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. USA 90:6701-6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan, M. P., and D. M. Knipe. 1985. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol. 5:957-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott, Williams, and Wilkins, Philadelphia, Pa.
- 51.Russell, A., J. Hendley, and D. Germain. 1999. Inhibitory effect of p21 in MCF-7 cells is overcome by its coordinated stabilization with D-type cyclins. Oncogene 18:6454-6459. [DOI] [PubMed] [Google Scholar]
- 52.Russell, A., M. A. Thompson, J. Hendley, L. Trute, J. Armes, and D. Germain. 1999. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene 18:1983-1991. [DOI] [PubMed] [Google Scholar]
- 53.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sears, A. E., I. W. Haliburton, B. Meignier, S. Silver, and B. Roizman. 1985. Herpes simplex virus 1 mutant deleted in the α22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 55:338-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seol, J. H., R. M. R. Feldman, W. Zachariae, A. Shevchenko, C. C. Correll, S. Lyapina, Y. Chi, M. Galova, J. Claypool, S. Sandmeyer, K. Nasmyth, A. Shevchenko, and R. J. Deshaies. 1999. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme cdc34. Genes Dev. 13:1614-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinclair, A. J., I. Palmero, G. Peters, and P. J. Farrell. 1994. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO. J. 13:3321-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song, B., J. J. Liu, K.-C. Yeh, and D. M. Knipe. 2000. Herpes simplex virus infection blocks events in the G1 phase of the cell cycle. Virology 267:326-334. [DOI] [PubMed] [Google Scholar]
- 58.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 59.Tyers, M., and A. R. Willems. 1999. One ring to rule a superfamily of E3 ubiquitin ligases. Science 284:603-604. [DOI] [PubMed] [Google Scholar]
- 60.Van Sant, C., P. Lopez, S. J. Advani, and B. Roizman. 2001. Role of cyclin D3 in the biology of herpes simplex virus 1 ICP0. J. Virol. 75:1888-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Sant, C., R. Hagglund, P. Lopez, and B. Roizman. 2001. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E3 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 98:8815-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Sant, C., Y. Kawaguchi, and B. Roizman. 1999. A single amino acid substitution in the cyclin D binding domain of the infected cell protein no. 0 abrogates the neuroinvasiveness of herpes simplex virus without affecting its ability to replicate. Proc. Natl. Acad. Sci. USA 96:8184-8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu, Z.-K., J. L. M. Gervais, and H. Zhang. 1998. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. USA 95:11324-11329. [DOI] [PMC free article] [PubMed] [Google Scholar]