

Abstract
In the molecule of the title compound, C15H15ClN2O3S, the S atom adopts a distorted tetrahedral coordination geometry with two O atoms, one N atom of the amide group and one C atom of the aromatic ring. An intramolecular O—H⋯N hydrogen bond results in the formation of a planar six-membered ring, which is oriented with respect to the adjacent aromatic ring at a dihedral angle of 3.38 (11)°. Thus, the two rings are nearly coplanar. In the crystal structure, intermolecular N—H⋯O hydrogen bonds link the molecules into centrosymmetric dimers.
Related literature
For general background, see: Supuran & Scozzafava (2001 ▶); Chohan & Shad (2007 ▶). For related literature, see: Chohan et al. (2008 ▶); Shad et al. (2008 ▶); Tahir et al. (2008 ▶); Li (2006 ▶).
Experimental
Crystal data
C15H15ClN2O3S
M r = 338.80
Monoclinic,
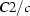
a = 21.069 (2) Å
b = 4.8125 (6) Å
c = 30.838 (3) Å
β = 99.942 (9)°
V = 3079.9 (6) Å3
Z = 8
Mo Kα radiation
μ = 0.40 mm−1
T = 296 (2) K
0.22 × 0.18 × 0.14 mm
Data collection
Bruker Kappa APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005 ▶) T min = 0.920, T max = 0.940
16002 measured reflections
4067 independent reflections
1851 reflections with I > 3σ(I)
R int = 0.060
Refinement
R[F 2 > 2σ(F 2)] = 0.046
wR(F 2) = 0.185
S = 1.02
4067 reflections
206 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.36 e Å−3
Δρmin = −0.28 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: APEX2; data reduction: SAINT (Bruker, 2007 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶) and PLATON (Spek, 2003 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶) and PLATON.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808005084/hk2428sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808005084/hk2428Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected geometric parameters (Å, °).
| S1—O2 | 1.430 (2) |
| S1—O3 | 1.423 (3) |
| S1—N2 | 1.616 (3) |
| S1—C13 | 1.752 (3) |
| O2—S1—O3 | 118.15 (14) |
| O2—S1—N2 | 105.67 (16) |
| O2—S1—C13 | 109.21 (15) |
| O3—S1—N2 | 108.20 (17) |
| O3—S1—C13 | 108.29 (15) |
| N2—S1—C13 | 106.76 (15) |
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯N1 | 0.82 | 1.83 | 2.554 (4) | 147.00 |
| N2—H2⋯O2i | 0.77 (4) | 2.30 (4) | 3.016 (4) | 156 (4) |
Symmetry code: (i) 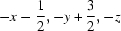 .
.
Acknowledgments
The authors acknowledge the Higher Education Commission, Islamabad, Pakistan, for funding the purchase of the diffractometer.
supplementary crystallographic information
Comment
The significance of sulfonamide was realised when sulfanilamide was first time reported as antibacterial drug. Later on, many sulfanilamide derivatives were synthesized, characterized and tested for antibacterial, anti-tumour, anti-carbonic anhydrase (Supuran & Scozzafava 2001), diuretic, hypoglycemic, anti-thyroid or protease inhibitory activity. Thus, sulfanilamide is performing a leading role for the development and expansion of all other types of medicinally important sulfonamides (Chohan & Shad, 2007).
The title compound, (I), is a reaction product of 5-chlorosalicylaldehyde and 4-(2-aminoethyl)benzenesulfonamide. The 4-(2-aminoethyl)benzenesulfonamide moiety is present in 4-[2-(3-ethyl-4-methyl-2-oxo-3-pyrrolidine-1-carboxamido)- ethyl]benzenesulfonamide (Li, 2006) and 5-chlorosalicylaldehyde exists in 4-[(5-chloro-2-hydroxybenzylidene)amino]-N-(3,4-dimethylisoxazol-5-yl)benzene- sulfonamide (Chohan et al., 2008). In continuation of synthesizing Schiff base ligands of substituted halogen salicylaldehydes and various sulfonamides (Chohan et al., 2008; Shad et al., 2008; Tahir et al., 2008), we report herein the crystal structure of the title compound, (I).
In the molecule of (I), S1 atom has a distorted tetrahedral coordination completed by the two O atoms, one N atom of amide group and one C atom of the adjacent aromatic ring B (C10—C15) (Table 1, Fig. 1). The two aromatic rings A (C1—C6) and B are connected by C=N—C—C group in a zigzag way, in which they are oriented at a dihedral angle of A/B = 23.95 (18)°. The intramolecular O—H···N hydrogen bond (Table 2) results in the formation of a planar six-membered ring C (O1/H1/N1/C7/C1/C2), which is oriented with respect to the aromatic rings at dihedral angles of A/C = 3.38 (11)° and B/C = 22.33 (13)°. So, rings A and C are also nearly coplanar.
In (I), C7—N1 [1.278 (5) Å] bond has double bond character. The C2=O1 [1.343 (5) Å] bond is a little longer than the corresponding value [1.328 (4) Å], as reported by Chohan et al. (2008). All other bonds in 5-chlorosalicylaldehyde moiety remain nearly same. The bond angles around S1 atom of sulfonamide group are a little smaller, having the range of 106.76 (15)° - 118.15 (14)°, compared to the values, in the range of 105.91 (13)°-119.68 (12)°, as reported by Li (2006).
In the crystal structure, intermolecular N—H···O hydrogen bonds (Table 2) link the molecules into centrosymmetric dimers (Fig. 2), in which they seem to be effective in the stabilization of the structure.
Experimental
For the preparation of the title compound, an ethanol solution (15 ml) of 4-(2-aminoethyl)benzenesulfonamide (400.5 mg, 2 mmol) was added to an ethanol solution (10 ml) of 5-chlorosalicylaldehyde (313.1 mg, 2 mmol). The reaction mixture was refluxed for 3 h. The colour of the solution gradually changed from colorless to greenish yellow. The solution was cooled to room temperature, filtered and the volume was reduced to about one-third on the rotary evaporator. It was allowed to stand for 10 d, bright yellow crystals of the title compound were obtained (m.p. 441 K).
Refinement
H atoms (for NH2) were located in a difference synthesis and refined isotropically [N—H = 0.77 (4) and 0.78 (4) Å; Uiso(H) = 0.0767 and 0.0613 Å2]. The remaining H atoms were positioned geometrically, with O—H = 0.82 Å (for OH) and C—H = 0.93 and 0.97 Å for aromatic and methylene H atoms, respectively, and constrained to ride on their parent atoms, with Uiso(H) = xUeq(C,O), where x = 1.5 for OH H and x = 1.2 for all other H atoms.
Figures
Fig. 1.

The molecular structure of the title molecule, with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

A packing diagram of (I). Hydrogen bonds are shown as dashed lines.
Crystal data
| C15H15ClN2O3S | F000 = 1408 |
| Mr = 338.80 | Dx = 1.461 Mg m−3 |
| Monoclinic, C2/c | Melting point: 497 K |
| Hall symbol: -C 2yc | Mo Kα radiation λ = 0.71073 Å |
| a = 21.069 (2) Å | Cell parameters from 2163 reflections |
| b = 4.8125 (6) Å | θ = 1.3–29.2º |
| c = 30.838 (3) Å | µ = 0.40 mm−1 |
| β = 99.942 (9)º | T = 296 (2) K |
| V = 3079.9 (6) Å3 | Prismatic, yellow |
| Z = 8 | 0.22 × 0.18 × 0.14 mm |
Data collection
| Bruker KappaAPEXII CCD diffractometer | 4067 independent reflections |
| Radiation source: fine-focus sealed tube | 1851 reflections with I > 3σ(I) |
| Monochromator: graphite | Rint = 0.060 |
| Detector resolution: 7.40 pixels mm-1 | θmax = 29.2º |
| T = 296(2) K | θmin = 1.3º |
| ω scans | h = −28→28 |
| Absorption correction: multi-scan(SADABS; Bruker, 2005) | k = −6→5 |
| Tmin = 0.920, Tmax = 0.940 | l = −41→42 |
| 16002 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.047 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.185 | w = 1/[σ2(Fo2) + (0.0.0962P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 4067 reflections | Δρmax = 0.36 e Å−3 |
| 206 parameters | Δρmin = −0.28 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All su's are estimated from the variances of the (full) variance-covariance matrix. The cell e.s.d.'s are taken into account in the estimation of distances, angles and torsion angles |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.32293 (6) | 0.3434 (3) | 0.35291 (5) | 0.1046 (5) | |
| S1 | −0.14925 (4) | 0.84474 (16) | −0.01326 (3) | 0.0411 (3) | |
| O1 | 0.07158 (14) | −0.1064 (6) | 0.27525 (10) | 0.0724 (11) | |
| O2 | −0.19990 (11) | 1.0016 (4) | 0.00047 (8) | 0.0535 (9) | |
| O3 | −0.10664 (12) | 0.9849 (5) | −0.03704 (8) | 0.0604 (9) | |
| N1 | 0.06373 (16) | 0.2662 (6) | 0.21557 (9) | 0.0566 (10) | |
| N2 | −0.18450 (17) | 0.5984 (6) | −0.04399 (11) | 0.0510 (11) | |
| C1 | 0.15618 (18) | 0.2192 (8) | 0.27152 (11) | 0.0494 (11) | |
| C2 | 0.12970 (19) | −0.0013 (8) | 0.29194 (12) | 0.0526 (11) | |
| C3 | 0.1641 (2) | −0.1103 (9) | 0.33014 (13) | 0.0687 (16) | |
| C4 | 0.2227 (2) | −0.0078 (9) | 0.34865 (13) | 0.0677 (16) | |
| C5 | 0.24889 (19) | 0.2059 (9) | 0.32861 (14) | 0.0663 (14) | |
| C6 | 0.2167 (2) | 0.3195 (9) | 0.29034 (13) | 0.0655 (14) | |
| C7 | 0.1197 (2) | 0.3508 (8) | 0.23331 (12) | 0.0590 (12) | |
| C8 | 0.0272 (2) | 0.4227 (8) | 0.17939 (12) | 0.0622 (14) | |
| C9 | 0.0102 (2) | 0.2436 (8) | 0.13928 (12) | 0.0653 (14) | |
| C10 | −0.02942 (19) | 0.3998 (7) | 0.10209 (11) | 0.0501 (11) | |
| C11 | −0.09656 (19) | 0.3851 (7) | 0.09454 (12) | 0.0556 (12) | |
| C12 | −0.13287 (17) | 0.5259 (7) | 0.06036 (11) | 0.0504 (11) | |
| C13 | −0.10314 (14) | 0.6854 (6) | 0.03276 (10) | 0.0355 (9) | |
| C14 | −0.03664 (16) | 0.7064 (8) | 0.03996 (12) | 0.0538 (11) | |
| C15 | −0.00076 (18) | 0.5662 (9) | 0.07457 (13) | 0.0625 (13) | |
| H1 | 0.05445 | −0.00964 | 0.25463 | 0.1087* | |
| H2 | −0.212 (2) | 0.526 (9) | −0.0349 (15) | 0.0767* | |
| H2A | −0.160 (2) | 0.495 (9) | −0.0511 (14) | 0.0613* | |
| H3 | 0.14678 | −0.25811 | 0.34366 | 0.0825* | |
| H4 | 0.24468 | −0.08276 | 0.37477 | 0.0812* | |
| H6 | 0.23528 | 0.46403 | 0.27690 | 0.0786* | |
| H7 | 0.13739 | 0.50280 | 0.22110 | 0.0710* | |
| H8A | 0.05252 | 0.58026 | 0.17261 | 0.0746* | |
| H8B | −0.01190 | 0.49318 | 0.18800 | 0.0746* | |
| H9A | 0.04945 | 0.17819 | 0.13017 | 0.0782* | |
| H9B | −0.01370 | 0.08266 | 0.14642 | 0.0782* | |
| H11 | −0.11706 | 0.27761 | 0.11306 | 0.0667* | |
| H12 | −0.17758 | 0.51368 | 0.05583 | 0.0607* | |
| H14 | −0.01635 | 0.81493 | 0.02147 | 0.0647* | |
| H15 | 0.04386 | 0.58389 | 0.07956 | 0.0747* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0648 (7) | 0.1294 (11) | 0.1019 (10) | 0.0029 (8) | −0.0357 (7) | −0.0252 (8) |
| S1 | 0.0365 (4) | 0.0370 (4) | 0.0461 (5) | −0.0006 (4) | −0.0030 (3) | 0.0111 (4) |
| O1 | 0.0634 (17) | 0.076 (2) | 0.0685 (19) | −0.0105 (16) | −0.0151 (14) | 0.0163 (14) |
| O2 | 0.0434 (14) | 0.0394 (12) | 0.0741 (18) | 0.0078 (11) | 0.0002 (13) | 0.0034 (12) |
| O3 | 0.0485 (14) | 0.0685 (15) | 0.0621 (16) | −0.0062 (14) | 0.0036 (13) | 0.0313 (13) |
| N1 | 0.0563 (18) | 0.0628 (18) | 0.0439 (17) | 0.0072 (17) | −0.0104 (15) | 0.0043 (14) |
| N2 | 0.054 (2) | 0.0502 (19) | 0.0440 (18) | −0.0025 (15) | −0.0054 (15) | 0.0014 (14) |
| C1 | 0.0497 (19) | 0.058 (2) | 0.0367 (18) | 0.0091 (18) | −0.0034 (16) | −0.0007 (16) |
| C2 | 0.057 (2) | 0.057 (2) | 0.0391 (19) | 0.012 (2) | −0.0050 (17) | −0.0024 (16) |
| C3 | 0.078 (3) | 0.069 (3) | 0.053 (2) | 0.012 (2) | −0.006 (2) | 0.010 (2) |
| C4 | 0.074 (3) | 0.076 (3) | 0.044 (2) | 0.024 (3) | −0.015 (2) | −0.002 (2) |
| C5 | 0.051 (2) | 0.086 (3) | 0.054 (2) | 0.015 (2) | −0.0134 (19) | −0.023 (2) |
| C6 | 0.057 (2) | 0.081 (3) | 0.053 (2) | 0.003 (2) | −0.0063 (19) | 0.001 (2) |
| C7 | 0.061 (2) | 0.066 (2) | 0.045 (2) | 0.001 (2) | −0.0047 (18) | 0.0111 (18) |
| C8 | 0.069 (3) | 0.055 (2) | 0.055 (2) | 0.014 (2) | −0.011 (2) | 0.0057 (18) |
| C9 | 0.083 (3) | 0.059 (2) | 0.045 (2) | 0.021 (2) | −0.014 (2) | 0.0039 (18) |
| C10 | 0.062 (2) | 0.046 (2) | 0.0371 (18) | 0.0077 (19) | −0.0063 (17) | 0.0017 (15) |
| C11 | 0.058 (2) | 0.061 (2) | 0.045 (2) | −0.005 (2) | 0.0013 (18) | 0.0167 (17) |
| C12 | 0.0418 (19) | 0.061 (2) | 0.045 (2) | −0.0051 (18) | −0.0017 (16) | 0.0101 (17) |
| C13 | 0.0348 (15) | 0.0331 (16) | 0.0366 (17) | −0.0014 (14) | 0.0003 (13) | 0.0030 (13) |
| C14 | 0.0354 (17) | 0.070 (2) | 0.054 (2) | −0.0030 (19) | 0.0025 (17) | 0.0195 (19) |
| C15 | 0.0372 (18) | 0.088 (3) | 0.058 (2) | 0.009 (2) | −0.0039 (17) | 0.012 (2) |
Geometric parameters (Å, °)
| Cl1—C5 | 1.741 (4) | C9—C10 | 1.499 (5) |
| S1—O2 | 1.430 (2) | C10—C15 | 1.380 (5) |
| S1—O3 | 1.423 (3) | C10—C11 | 1.395 (6) |
| S1—N2 | 1.616 (3) | C11—C12 | 1.370 (5) |
| S1—C13 | 1.752 (3) | C12—C13 | 1.375 (5) |
| O1—C2 | 1.343 (5) | C13—C14 | 1.384 (5) |
| O1—H1 | 0.8200 | C14—C15 | 1.374 (5) |
| N1—C7 | 1.278 (5) | C3—H3 | 0.9300 |
| N1—C8 | 1.452 (5) | C4—H4 | 0.9300 |
| N2—H2 | 0.77 (4) | C6—H6 | 0.9300 |
| N2—H2A | 0.78 (4) | C7—H7 | 0.9300 |
| C1—C7 | 1.438 (5) | C8—H8A | 0.9700 |
| C1—C2 | 1.398 (5) | C8—H8B | 0.9700 |
| C1—C6 | 1.393 (6) | C9—H9A | 0.9700 |
| C2—C3 | 1.376 (6) | C9—H9B | 0.9700 |
| C3—C4 | 1.360 (6) | C11—H11 | 0.9300 |
| C4—C5 | 1.365 (6) | C12—H12 | 0.9300 |
| C5—C6 | 1.369 (6) | C14—H14 | 0.9300 |
| C8—C9 | 1.499 (5) | C15—H15 | 0.9300 |
| O2—S1—O3 | 118.15 (14) | S1—C13—C12 | 119.8 (2) |
| O2—S1—N2 | 105.67 (16) | C12—C13—C14 | 119.9 (3) |
| O2—S1—C13 | 109.21 (15) | S1—C13—C14 | 120.2 (2) |
| O3—S1—N2 | 108.20 (17) | C13—C14—C15 | 119.7 (3) |
| O3—S1—C13 | 108.29 (15) | C10—C15—C14 | 121.5 (4) |
| N2—S1—C13 | 106.76 (15) | C2—C3—H3 | 119.00 |
| C2—O1—H1 | 109.00 | C4—C3—H3 | 119.00 |
| C7—N1—C8 | 119.4 (3) | C3—C4—H4 | 120.00 |
| S1—N2—H2 | 115 (3) | C5—C4—H4 | 120.00 |
| S1—N2—H2A | 112 (3) | C1—C6—H6 | 120.00 |
| H2—N2—H2A | 113 (5) | C5—C6—H6 | 120.00 |
| C2—C1—C7 | 120.3 (3) | N1—C7—H7 | 119.00 |
| C6—C1—C7 | 120.7 (4) | C1—C7—H7 | 119.00 |
| C2—C1—C6 | 118.9 (3) | N1—C8—H8A | 109.00 |
| O1—C2—C1 | 121.4 (3) | N1—C8—H8B | 110.00 |
| O1—C2—C3 | 119.7 (4) | C9—C8—H8A | 109.00 |
| C1—C2—C3 | 118.9 (4) | C9—C8—H8B | 110.00 |
| C2—C3—C4 | 121.8 (4) | H8A—C8—H8B | 108.00 |
| C3—C4—C5 | 119.5 (4) | C8—C9—H9A | 109.00 |
| Cl1—C5—C4 | 119.6 (3) | C8—C9—H9B | 109.00 |
| Cl1—C5—C6 | 119.5 (3) | C10—C9—H9A | 109.00 |
| C4—C5—C6 | 120.8 (4) | C10—C9—H9B | 109.00 |
| C1—C6—C5 | 120.2 (4) | H9A—C9—H9B | 108.00 |
| N1—C7—C1 | 122.3 (4) | C10—C11—H11 | 119.00 |
| N1—C8—C9 | 110.8 (3) | C12—C11—H11 | 119.00 |
| C8—C9—C10 | 111.4 (3) | C11—C12—H12 | 120.00 |
| C9—C10—C15 | 121.1 (4) | C13—C12—H12 | 120.00 |
| C11—C10—C15 | 117.8 (3) | C13—C14—H14 | 120.00 |
| C9—C10—C11 | 121.1 (3) | C15—C14—H14 | 120.00 |
| C10—C11—C12 | 121.3 (3) | C10—C15—H15 | 119.00 |
| C11—C12—C13 | 119.9 (3) | C14—C15—H15 | 119.00 |
| O2—S1—C13—C12 | 52.8 (3) | C2—C3—C4—C5 | −1.1 (6) |
| O2—S1—C13—C14 | −131.0 (3) | C3—C4—C5—Cl1 | 178.4 (3) |
| O3—S1—C13—C12 | −177.3 (3) | C3—C4—C5—C6 | 0.5 (6) |
| O3—S1—C13—C14 | −1.1 (3) | Cl1—C5—C6—C1 | −177.3 (3) |
| N2—S1—C13—C12 | −61.0 (3) | C4—C5—C6—C1 | 0.6 (6) |
| N2—S1—C13—C14 | 115.2 (3) | N1—C8—C9—C10 | 178.1 (3) |
| C8—N1—C7—C1 | 174.7 (3) | C8—C9—C10—C11 | −95.0 (4) |
| C7—N1—C8—C9 | 122.1 (4) | C8—C9—C10—C15 | 84.1 (5) |
| C6—C1—C2—O1 | −180.0 (4) | C9—C10—C11—C12 | −179.5 (3) |
| C6—C1—C2—C3 | 0.5 (6) | C15—C10—C11—C12 | 1.5 (5) |
| C7—C1—C2—O1 | 3.9 (6) | C9—C10—C15—C14 | 178.9 (4) |
| C7—C1—C2—C3 | −175.7 (4) | C11—C10—C15—C14 | −2.0 (6) |
| C2—C1—C6—C5 | −1.1 (6) | C10—C11—C12—C13 | 0.0 (5) |
| C7—C1—C6—C5 | 175.1 (4) | C11—C12—C13—S1 | 175.4 (3) |
| C2—C1—C7—N1 | −3.0 (6) | C11—C12—C13—C14 | −0.9 (5) |
| C6—C1—C7—N1 | −179.1 (4) | S1—C13—C14—C15 | −175.9 (3) |
| O1—C2—C3—C4 | −179.0 (4) | C12—C13—C14—C15 | 0.3 (5) |
| C1—C2—C3—C4 | 0.6 (6) | C13—C14—C15—C10 | 1.2 (6) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1 | 0.8200 | 1.8300 | 2.554 (4) | 147.00 |
| N2—H2···O2i | 0.77 (4) | 2.30 (4) | 3.016 (4) | 156 (4) |
Symmetry codes: (i) −x−1/2, −y+3/2, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2428).
References
- Bruker (2005). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2007). APEX2 (Version 1.27) and SAINT (Version 7.12a). Bruker AXS Inc., Madison, Wisconsin, USA.
- Chohan, Z. H. & Shad, H. A. (2007). J. Enz. Inhib. Med. Chem. http://dx.doi.org/10.1080/14756360701585692.
- Chohan, Z. H., Tahir, M. N., Shad, H. A. & Khan, I. U. (2008). Acta Cryst. E64, o648. [DOI] [PMC free article] [PubMed]
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Li, X. (2006). Acta Cryst. E62, o3019–o3020.
- Shad, H. A., Chohan, Z. H., Tahir, M. N. & Khan, I. U. (2008). Acta Cryst. E64, o635. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Supuran, C. T. & Scozzafava, A. (2001). Curr. Med. Chem.-Imm. Endoc. Metab. Agents, 1, 61–97.
- Tahir, M. N., Chohan, Z. H., Shad, H. A. & Khan, I. U. (2008). Acta Cryst. E64, o720. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808005084/hk2428sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808005084/hk2428Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report