

Abstract
The structure of the title compound, C13H10ClNO, resembles those of N-phenylbenzamide, N-(2-chlorophenyl)benzamide and other benzanilides, with similar bond parameters. The amide group –NHCO– makes a dihedral angle of 29.95 (9)° with the benzoyl ring, while the benzoyl and aniline rings form a dihedral angle of 60.76 (3)°. The structure shows both intra- and intermolecular hydrogen bonding. The molecules are linked by N—H⋯O hydrogen bonds into chains running along the [100] direction.
Related literature
For related literature, see: Gowda et al. (2003 ▶, 2007 ▶, 2008 ▶).
Experimental
Crystal data
C13H10ClNO
M r = 231.67
Triclinic,
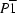
a = 5.3789 (1) Å
b = 7.8501 (2) Å
c = 13.6318 (4) Å
α = 106.509 (2)°
β = 98.380 (2)°
γ = 90.631 (2)°
V = 545.15 (2) Å3
Z = 2
Mo Kα radiation
μ = 0.33 mm−1
T = 295 (2) K
0.52 × 0.25 × 0.08 mm
Data collection
Oxford Xcalibur diffractometer
Absorption correction: analytical [CrysAlis RED (Oxford Diffraction, 2007 ▶), using a multifaceted crystal model based on expressions derived by Clark & Reid (1995 ▶)] T min = 0.852, T max = 0.975
23656 measured reflections
2087 independent reflections
1773 reflections with I > 2σ(I)
R int = 0.026
Refinement
R[F 2 > 2σ(F 2)] = 0.032
wR(F 2) = 0.091
S = 1.08
2087 reflections
148 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.19 e Å−3
Δρmin = −0.23 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2007 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2007 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and DIAMOND (Brandenburg, 2002 ▶); software used to prepare material for publication: SHELXL97, PLATON (Spek, 2003 ▶) and WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808008155/dn2327sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808008155/dn2327Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C9—H9⋯O1 | 0.93 | 2.43 | 2.9090 (17) | 112 |
| N1—H1N⋯O1i | 0.845 (16) | 2.390 (16) | 3.1710 (15) | 154.0 (15) |
| C13—H13⋯O1i | 0.93 | 2.58 | 3.2507 (11) | 129 |
Symmetry code: (i) 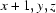 .
.
Acknowledgments
MT and JK thank the Grant Agency of the Slovak Republic (grant No. VEGA 1/0817/08) and the Structural Funds, Interreg IIIA, for financial support in purchasing the diffractometer.
supplementary crystallographic information
Comment
In the present work, the structure of N-(4-chlorophenyl)benzamide (N4CPBA) has been determined to study the effect of substituents on the structures of benzanilides (Gowda et al., 2003, 2007, 2008).
The structure of N4CPBA (Fig.1) is similar to those of N-(phenyl)benzamide, N-(2-chlorophenyl)benzamide, N-(3-chlorophenyl)benzamide and N-(4-methylphenyl)benzamide and other benzanilides (Gowda et al., 2003, 2007, 2008). The amide group –NHCO– forms dihedral angle of 29.95 (9)° with the benzoyl ring, while the two benzene rings (benzoyl and aniline rings) form dihedral angle of 60.76 (3)°. Part of the structure of N4CPBA as viewed down the b axis and showing infinite molecular chains in the [100] direction is shown in Fig. 2. The chains are generated by the intermolecular N—H···O hydrogen bonds (Table 1).
Experimental
The title compound was prepared according to the literature method (Gowda et al., 2003). The purity of the compound was checked by determining its melting point. It was characterized by recording its infrared and NMR spectra. Single crystals of the title compound were obtained from an ethanolic solution and used for X-ray diffraction studies at room temperature.
Refinement
H atoms attached to C atoms were placed in calculated positions and subsequently treated as riding with C—H distance 0.93 Å. H atom of the amide group was refined with the N—H distance restrained to 0.86 (4) Å. The Uiso(H) values were set at 1.2 Ueq(C,N).
Figures
Fig. 1.

Molecular structure of the title compound showing the atom labelling scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms are represented as small spheres of arbitrary radii. Intramolecular hydrogen bond C9—H9···O1 is shown by dashed line.
Fig. 2.

Part of the crystal structure of the title compound viewed down the b axis and showing infinite molecular chains in the[100] direction. H atoms not involved in intermolecular bonding have been omitted. [Symmetry code: (i) x + 1, y, z]
Crystal data
| C13H10ClNO | Z = 2 |
| Mr = 231.67 | F000 = 240 |
| Triclinic, P1 | Dx = 1.411 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation λ = 0.71073 Å |
| a = 5.3789 (1) Å | Cell parameters from 13860 reflections |
| b = 7.8501 (2) Å | θ = 3.1–29.3º |
| c = 13.6318 (4) Å | µ = 0.33 mm−1 |
| α = 106.509 (2)º | T = 295 (2) K |
| β = 98.380 (2)º | Block, colorless |
| γ = 90.631 (2)º | 0.52 × 0.25 × 0.08 mm |
| V = 545.15 (2) Å3 |
Data collection
| Oxford Xcalibur diffractometer | 2087 independent reflections |
| Radiation source: fine-focus sealed tube | 1773 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.026 |
| Detector resolution: 10.434 pixels mm-1 | θmax = 25.8º |
| T = 295(2) K | θmin = 5.6º |
| φ scans, and ω scans with κ offsets | h = −6→6 |
| Absorption correction: analytical[CrysAlis RED (Oxford Diffraction, 2007). Analytical absorption correction using a multifaceted crystal model based on expressions derived by Clark & Reid (1995)] | k = −9→9 |
| Tmin = 0.852, Tmax = 0.975 | l = −16→16 |
| 23656 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.032 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.091 | w = 1/[σ2(Fo2) + (0.0494P)2 + 0.0939P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.08 | (Δ/σ)max = 0.001 |
| 2087 reflections | Δρmax = 0.19 e Å−3 |
| 148 parameters | Δρmin = −0.23 e Å−3 |
| 1 restraint | Extinction correction: none |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.3126 (2) | 0.77177 (16) | 0.02982 (9) | 0.0381 (3) | |
| H1N | 0.443 (3) | 0.757 (2) | 0.0010 (13) | 0.046* | |
| O1 | −0.11350 (19) | 0.74813 (17) | 0.00041 (8) | 0.0563 (3) | |
| C1 | 0.0870 (2) | 0.71549 (18) | −0.03078 (10) | 0.0370 (3) | |
| C2 | 0.0976 (2) | 0.61312 (17) | −0.14032 (10) | 0.0343 (3) | |
| C3 | −0.1018 (2) | 0.62387 (19) | −0.21496 (11) | 0.0396 (3) | |
| H3 | −0.2346 | 0.6941 | −0.1956 | 0.047* | |
| C4 | −0.1045 (3) | 0.5314 (2) | −0.31747 (11) | 0.0450 (3) | |
| H4 | −0.2376 | 0.5409 | −0.3671 | 0.054* | |
| C5 | 0.0893 (3) | 0.4249 (2) | −0.34662 (11) | 0.0460 (4) | |
| H5 | 0.0871 | 0.3621 | −0.4158 | 0.055* | |
| C6 | 0.2867 (3) | 0.41171 (19) | −0.27281 (12) | 0.0452 (3) | |
| H6 | 0.4167 | 0.3387 | −0.2924 | 0.054* | |
| C7 | 0.2930 (3) | 0.50593 (18) | −0.17026 (11) | 0.0392 (3) | |
| H7 | 0.4281 | 0.4977 | −0.1211 | 0.047* | |
| C8 | 0.3531 (2) | 0.87653 (17) | 0.13439 (10) | 0.0337 (3) | |
| C9 | 0.1943 (3) | 0.86476 (19) | 0.20396 (10) | 0.0396 (3) | |
| H9 | 0.0509 | 0.7884 | 0.1818 | 0.048* | |
| C10 | 0.2490 (3) | 0.96646 (19) | 0.30619 (11) | 0.0415 (3) | |
| H10 | 0.1426 | 0.9588 | 0.353 | 0.05* | |
| C11 | 0.4617 (3) | 1.07920 (18) | 0.33843 (10) | 0.0396 (3) | |
| C12 | 0.6211 (3) | 1.09201 (19) | 0.27047 (11) | 0.0419 (3) | |
| H12 | 0.7643 | 1.1685 | 0.293 | 0.05* | |
| C13 | 0.56679 (8) | 0.99040 (5) | 0.16848 (3) | 0.0391 (3) | |
| H13 | 0.6744 | 0.9983 | 0.1222 | 0.047* | |
| Cl1 | 0.53006 (8) | 1.20876 (5) | 0.46690 (3) | 0.06227 (17) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0320 (6) | 0.0455 (7) | 0.0326 (6) | 0.0003 (5) | 0.0068 (5) | 0.0037 (5) |
| O1 | 0.0336 (5) | 0.0849 (8) | 0.0394 (6) | 0.0060 (5) | 0.0073 (4) | −0.0005 (5) |
| C1 | 0.0337 (7) | 0.0404 (7) | 0.0347 (7) | 0.0024 (5) | 0.0045 (5) | 0.0076 (6) |
| C2 | 0.0328 (7) | 0.0340 (7) | 0.0345 (7) | −0.0022 (5) | 0.0060 (5) | 0.0069 (5) |
| C3 | 0.0328 (7) | 0.0422 (7) | 0.0400 (7) | 0.0032 (5) | 0.0043 (5) | 0.0066 (6) |
| C4 | 0.0402 (8) | 0.0505 (8) | 0.0379 (7) | −0.0029 (6) | −0.0035 (6) | 0.0075 (6) |
| C5 | 0.0487 (8) | 0.0474 (8) | 0.0342 (7) | −0.0054 (6) | 0.0074 (6) | −0.0009 (6) |
| C6 | 0.0394 (8) | 0.0436 (8) | 0.0471 (8) | 0.0043 (6) | 0.0117 (6) | 0.0020 (6) |
| C7 | 0.0335 (7) | 0.0412 (7) | 0.0394 (7) | 0.0023 (5) | 0.0026 (5) | 0.0077 (6) |
| C8 | 0.0325 (6) | 0.0343 (7) | 0.0323 (6) | 0.0043 (5) | 0.0039 (5) | 0.0067 (5) |
| C9 | 0.0343 (7) | 0.0451 (8) | 0.0369 (7) | −0.0042 (6) | 0.0026 (5) | 0.0097 (6) |
| C10 | 0.0404 (7) | 0.0510 (8) | 0.0333 (7) | 0.0031 (6) | 0.0084 (6) | 0.0113 (6) |
| C11 | 0.0442 (8) | 0.0378 (7) | 0.0317 (7) | 0.0065 (6) | 0.0004 (6) | 0.0043 (5) |
| C12 | 0.0369 (7) | 0.0385 (7) | 0.0441 (8) | −0.0035 (6) | 0.0005 (6) | 0.0051 (6) |
| C13 | 0.0347 (7) | 0.0416 (7) | 0.0401 (7) | 0.0007 (5) | 0.0091 (6) | 0.0089 (6) |
| Cl1 | 0.0764 (3) | 0.0615 (3) | 0.0352 (2) | −0.0009 (2) | −0.00138 (18) | −0.00280 (18) |
Geometric parameters (Å, °)
| N1—C1 | 1.3560 (18) | C6—H6 | 0.93 |
| N1—C8 | 1.4125 (17) | C7—H7 | 0.93 |
| N1—H1N | 0.845 (16) | C8—C13 | 1.3862 (13) |
| O1—C1 | 1.2196 (16) | C8—C9 | 1.3862 (18) |
| C1—C2 | 1.4909 (18) | C9—C10 | 1.3817 (19) |
| C2—C3 | 1.3876 (19) | C9—H9 | 0.93 |
| C2—C7 | 1.3885 (19) | C10—C11 | 1.377 (2) |
| C3—C4 | 1.377 (2) | C10—H10 | 0.93 |
| C3—H3 | 0.93 | C11—C12 | 1.373 (2) |
| C4—C5 | 1.376 (2) | C11—Cl1 | 1.7402 (14) |
| C4—H4 | 0.93 | C12—C13 | 1.3786 (15) |
| C5—C6 | 1.379 (2) | C12—H12 | 0.93 |
| C5—H5 | 0.93 | C13—H13 | 0.93 |
| C6—C7 | 1.379 (2) | ||
| C1—N1—C8 | 126.64 (11) | C6—C7—C2 | 120.02 (13) |
| C1—N1—H1N | 117.7 (11) | C6—C7—H7 | 120 |
| C8—N1—H1N | 115.0 (12) | C2—C7—H7 | 120 |
| O1—C1—N1 | 123.02 (12) | C13—C8—C9 | 119.42 (11) |
| O1—C1—C2 | 121.31 (12) | C13—C8—N1 | 117.74 (10) |
| N1—C1—C2 | 115.66 (11) | C9—C8—N1 | 122.80 (12) |
| C3—C2—C7 | 119.04 (12) | C10—C9—C8 | 120.05 (12) |
| C3—C2—C1 | 117.62 (11) | C10—C9—H9 | 120 |
| C7—C2—C1 | 123.33 (12) | C8—C9—H9 | 120 |
| C4—C3—C2 | 120.51 (12) | C11—C10—C9 | 119.65 (12) |
| C4—C3—H3 | 119.7 | C11—C10—H10 | 120.2 |
| C2—C3—H3 | 119.7 | C9—C10—H10 | 120.2 |
| C5—C4—C3 | 120.17 (13) | C12—C11—C10 | 120.97 (13) |
| C5—C4—H4 | 119.9 | C12—C11—Cl1 | 119.21 (11) |
| C3—C4—H4 | 119.9 | C10—C11—Cl1 | 119.82 (11) |
| C4—C5—C6 | 119.71 (13) | C11—C12—C13 | 119.40 (12) |
| C4—C5—H5 | 120.1 | C11—C12—H12 | 120.3 |
| C6—C5—H5 | 120.1 | C13—C12—H12 | 120.3 |
| C7—C6—C5 | 120.53 (13) | C12—C13—C8 | 120.52 (9) |
| C7—C6—H6 | 119.7 | C12—C13—H13 | 119.7 |
| C5—C6—H6 | 119.7 | C8—C13—H13 | 119.7 |
| C8—N1—C1—O1 | −1.4 (2) | C1—C2—C7—C6 | 178.39 (12) |
| C8—N1—C1—C2 | 177.27 (12) | C1—N1—C8—C13 | −149.06 (12) |
| O1—C1—C2—C3 | 28.33 (19) | C1—N1—C8—C9 | 33.4 (2) |
| N1—C1—C2—C3 | −150.35 (12) | C13—C8—C9—C10 | 0.24 (18) |
| O1—C1—C2—C7 | −150.27 (14) | N1—C8—C9—C10 | 177.75 (12) |
| N1—C1—C2—C7 | 31.06 (18) | C8—C9—C10—C11 | 0.0 (2) |
| C7—C2—C3—C4 | −0.8 (2) | C9—C10—C11—C12 | −0.1 (2) |
| C1—C2—C3—C4 | −179.47 (12) | C9—C10—C11—Cl1 | 179.53 (10) |
| C2—C3—C4—C5 | 1.0 (2) | C10—C11—C12—C13 | 0.0 (2) |
| C3—C4—C5—C6 | −0.3 (2) | Cl1—C11—C12—C13 | −179.61 (9) |
| C4—C5—C6—C7 | −0.7 (2) | C11—C12—C13—C8 | 0.18 (17) |
| C5—C6—C7—C2 | 1.0 (2) | C9—C8—C13—C12 | −0.32 (15) |
| C3—C2—C7—C6 | −0.2 (2) | N1—C8—C13—C12 | −177.95 (10) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C9—H9···O1 | 0.93 | 2.43 | 2.9090 (17) | 112 |
| N1—H1N···O1i | 0.845 (16) | 2.390 (16) | 3.1710 (15) | 154.0 (15) |
| C13—H13···O1i | 0.93 | 2.58 | 3.2507 (11) | 129 |
Symmetry codes: (i) x+1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: DN2327).
References
- Brandenburg, K. (2002). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Clark, R. C. & Reid, J. S. (1995). Acta Cryst. A51, 887–897.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Gowda, B. T., Jyothi, K., Paulus, H. & Fuess, H. (2003). Z. Naturforsch. Teil A, 58, 225–230.
- Gowda, B. T., Sowmya, B. P., Kožíšek, J., Tokarčík, M. & Fuess, H. (2007). Acta Cryst. E63, o2906. [DOI] [PMC free article] [PubMed]
- Gowda, B. T., Tokarčík, M., Kožíšek, J. & Sowmya, B. P. (2008). Acta Cryst. E64, o83. [DOI] [PMC free article] [PubMed]
- Oxford Diffraction (2007). CrysAlis CCD and CrysAlis RED Oxford Diffraction Ltd, Abingdon, Oxfordshire, England.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808008155/dn2327sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808008155/dn2327Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report