

Abstract
The crystal structure of the title compound, which is also known as vitamin B13 (systematic name: 2,6-dioxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid monohydrate), C5H4N2O4·H2O, was reported for the first time by Takusagawa & Shimada [Bull. Chem. Soc. Jpn (1973 ▶), 46, 2011–2019]. The present redetermination provides more precise values of the molecular geometry. The asymmetric unit comprises a planar diketo tautomer and a solvent water molecule. In the crystal structure, molecules are connected by O—H⋯O, N—H⋯O and C—H⋯O hydrogen bonds involving NH groups, two carbonyl O atoms and the solvent water molecule.
Related literature
For the previous structure determination, see: Takusagawa & Shimada (1973 ▶). For a general approach to the use of multiple hydrogen-bonding DNA/RNA nucleobases as potential supramolecular reagents, see: Portalone et al. (1999 ▶); Brunetti et al. (2000 ▶, 2002 ▶); Portalone & Colapietro (2007 ▶ and references therein). For the computation of ring patterns formed by hydrogen bonds in crystal structures, see: Etter et al. (1990 ▶); Bernstein et al. (1995 ▶); Motherwell et al. (1999 ▶).
Experimental
Crystal data
C5H4N2O4·H2O
M r = 174.12
Triclinic,
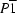
a = 5.89854 (14) Å
b = 6.92921 (15) Å
c = 9.59160 (18) Å
α = 74.6778 (12)°
β = 72.3232 (16)°
γ = 68.447 (2)°
V = 342.21 (1) Å3
Z = 2
Mo Kα radiation
μ = 0.15 mm−1
T = 298 (2) K
0.20 × 0.15 × 0.15 mm
Data collection
Oxford Diffraction Xcalibur S CCD diffractometer
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2006 ▶) T min = 0.977, T max = 0.985
64781 measured reflections
2340 independent reflections
2048 reflections with I > 2σ(I)
R int = 0.017
Refinement
R[F 2 > 2σ(F 2)] = 0.044
wR(F 2) = 0.133
S = 1.08
2340 reflections
133 parameters
All H-atom parameters refined
Δρmax = 0.19 e Å−3
Δρmin = −0.22 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2006 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2006 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP3 (Farrugia, 1997 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680800562X/kp2160sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680800562X/kp2160Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O4—H4⋯O5 | 0.89 (3) | 1.65 (3) | 2.5231 (11) | 166 (2) |
| N1—H1⋯O1i | 0.85 (2) | 2.03 (2) | 2.8824 (11) | 175.2 (19) |
| N3—H3⋯O2ii | 0.94 (2) | 1.87 (2) | 2.8112 (11) | 174.6 (18) |
| O5—H51⋯O2iii | 0.81 (3) | 2.00 (3) | 2.7786 (12) | 161 (3) |
| O5—H52⋯O3iv | 0.82 (3) | 1.98 (3) | 2.7787 (12) | 164 (2) |
| C5—H5⋯O4iii | 0.879 (19) | 2.740 (19) | 3.5922 (13) | 163.7 (15) |
Symmetry codes: (i) 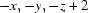 ; (ii)
; (ii) 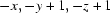 ; (iii)
; (iii) 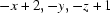 ; (iv)
; (iv) 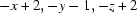 .
.
Acknowledgments
The author thanks MIUR (Rome) for financial support in 2006 of the project ‘X-ray diffractometry and spectrometry’.
supplementary crystallographic information
Comment
Orotic acid monohydrate, the only effective precursor in the biosynthesis of pyrimidine nucleobases, was determined some 35 years ago (Takusagawa & Shimada, 1973). In this study, 1488 unique reflections were collected at an ambient temperature by photographic techniques using Cu Kα radiation. 1344 of these, having values significantly above background, were estimated visually and no absorption correction was applied [µ(Cu Kα) = 15.4 cm-1]. The final refinement led to R = 0.058 with standard deviations of 0.005Å in C—C bond lengths, As a part of a more general study of multiple hydrogen-bonded DNA/RNA-nucleobases as potential supramolecular reagents (Brunetti et al., 2000, 2002; Portalone et al., 1999; Portalone & Colapietro, 2007), this paper reports a redetermination of the crystal structure of the title compound, (I), with greater precision and accuracy. The asymmetric unit of (I) (Fig. 1) comprises a planar diketo tautomer and a crystal water molecule. The analysis of the crystal packing of (I) shows five N—H···O and O—H···O intermolecular hydrogen bonds (Table 1) which link the molecules into planar layers (Fig. 2). Two kinds of inversion-related N—H···O hydrogen bonds with graph-set motifs R22(8) (Etter et al., 1990; Bernstein et al., 1995; Motherwell et al., 1999) (Table 1) are formed between NH groups and two carbonyl O atoms (O1i and O2ii) [symmetry code: (i) -x, -y, z + 2; (ii) -x, -y + 1, -z + 1] to give a zigzag chain. Each chain is joined by three O—H···O intermolecular hydrogen bonds to form rings of descriptor R44(12), R43(13) and R44(18) through a water molecule (O4—H4···O5, O5—H51···O2iii and O5—H52···O2iv; symmetry code: (iii) -x + 2, -y, -z + 1; (iv) -x + 2, -y - 1, -z + 2). The hydrogen-bonded two-dimensional array involves the C5—H5···O4iii intermolecular interaction.
Experimental
The title compound (0.1 mmol, Sigma Aldrich of 98% purity) was dissolved in water (9 ml) and heated under reflux for 2 h. After cooling the solution to ambient temperature, crystals suitable for single-crystal X-ray diffraction were grown by slow evaporation.
Refinement
All H atoms were found in a difference map and refined isotropically.
Figures
Fig. 1.

The molecular structure of (I), showing the atom-labelling scheme. Displacements ellipsoids are at the 50% probability level. Hydrogen bonding is indicated by dashed lines.
Fig. 2.

Crystal packing diagram of (I). All atoms are shown as small spheres of arbitrary radii. Hydrogen bonding is indicated by dashed lines.
Crystal data
| C5H4N2O4·H2O | Z = 2 |
| Mr = 174.12 | F000 = 180 |
| Triclinic, P1 | Dx = 1.690 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation λ = 0.71073 Å |
| a = 5.89854 (14) Å | Cell parameters from 64781 reflections |
| b = 6.92921 (15) Å | θ = 3.2–32.0º |
| c = 9.59160 (18) Å | µ = 0.15 mm−1 |
| α = 74.6778 (12)º | T = 298 (2) K |
| β = 72.3232 (16)º | Plate, colourless |
| γ = 68.447 (2)º | 0.20 × 0.15 × 0.15 mm |
| V = 342.207 (13) Å3 |
Data collection
| Oxford Diffraction Xcalibur S CCD diffractometer | 2340 independent reflections |
| Radiation source: Enhance (Mo) X-ray source | 2048 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.017 |
| Detector resolution: 16.0696 pixels mm-1 | θmax = 32.0º |
| T = 298(2) K | θmin = 3.2º |
| ω and φ scans | h = −8→8 |
| Absorption correction: multi-scan(CrysAlis RED; Oxford Diffraction, 2006) | k = −10→10 |
| Tmin = 0.977, Tmax = 0.985 | l = −14→14 |
| 64781 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.044 | All H-atom parameters refined |
| wR(F2) = 0.133 | w = 1/[σ2(Fo2) + (0.0846P)2 + 0.0376P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.08 | (Δ/σ)max < 0.001 |
| 2340 reflections | Δρmax = 0.19 e Å−3 |
| 133 parameters | Δρmin = −0.22 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Experimental. Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | −0.14403 (15) | 0.19593 (14) | 0.87497 (9) | 0.0437 (2) | |
| O2 | 0.33017 (14) | 0.40869 (13) | 0.43298 (8) | 0.0402 (2) | |
| O3 | 0.67729 (18) | −0.25757 (17) | 0.92057 (11) | 0.0575 (3) | |
| O4 | 0.94015 (16) | −0.17721 (14) | 0.70790 (10) | 0.0430 (2) | |
| H4 | 1.045 (5) | −0.276 (4) | 0.759 (3) | 0.072 (6)* | |
| N1 | 0.27952 (15) | 0.03091 (13) | 0.83018 (9) | 0.03108 (19) | |
| H1 | 0.248 (4) | −0.038 (3) | 0.918 (2) | 0.052 (4)* | |
| C2 | 0.06502 (18) | 0.17428 (15) | 0.79307 (10) | 0.0302 (2) | |
| N3 | 0.09988 (15) | 0.29328 (13) | 0.65310 (8) | 0.03059 (19) | |
| H3 | −0.050 (4) | 0.391 (3) | 0.630 (2) | 0.054 (5)* | |
| C4 | 0.32531 (17) | 0.28849 (14) | 0.55477 (10) | 0.0284 (2) | |
| C5 | 0.54286 (17) | 0.13666 (14) | 0.60218 (10) | 0.0295 (2) | |
| H5 | 0.689 (3) | 0.127 (3) | 0.540 (2) | 0.042 (4)* | |
| C6 | 0.51057 (17) | 0.01317 (13) | 0.73716 (9) | 0.02706 (19) | |
| C7 | 0.72006 (19) | −0.15575 (15) | 0.79814 (11) | 0.0325 (2) | |
| O5 | 1.28887 (17) | −0.45822 (16) | 0.81521 (11) | 0.0523 (3) | |
| H51 | 1.421 (5) | −0.460 (4) | 0.757 (3) | 0.080 (7)* | |
| H52 | 1.313 (5) | −0.528 (4) | 0.896 (3) | 0.082 (7)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0285 (4) | 0.0499 (5) | 0.0329 (4) | −0.0054 (3) | −0.0022 (3) | 0.0094 (3) |
| O2 | 0.0291 (4) | 0.0447 (4) | 0.0298 (4) | −0.0065 (3) | −0.0079 (3) | 0.0154 (3) |
| O3 | 0.0390 (5) | 0.0612 (6) | 0.0449 (5) | −0.0062 (4) | −0.0138 (4) | 0.0266 (4) |
| O4 | 0.0302 (4) | 0.0449 (4) | 0.0385 (4) | −0.0003 (3) | −0.0112 (3) | 0.0054 (3) |
| N1 | 0.0289 (4) | 0.0311 (4) | 0.0247 (3) | −0.0063 (3) | −0.0080 (3) | 0.0067 (3) |
| C2 | 0.0277 (4) | 0.0308 (4) | 0.0252 (4) | −0.0071 (3) | −0.0065 (3) | 0.0037 (3) |
| N3 | 0.0245 (4) | 0.0319 (4) | 0.0258 (3) | −0.0047 (3) | −0.0077 (3) | 0.0070 (3) |
| C4 | 0.0260 (4) | 0.0289 (4) | 0.0244 (4) | −0.0064 (3) | −0.0078 (3) | 0.0041 (3) |
| C5 | 0.0243 (4) | 0.0308 (4) | 0.0258 (4) | −0.0045 (3) | −0.0072 (3) | 0.0029 (3) |
| C6 | 0.0268 (4) | 0.0251 (4) | 0.0257 (4) | −0.0045 (3) | −0.0106 (3) | 0.0014 (3) |
| C7 | 0.0306 (4) | 0.0297 (4) | 0.0322 (4) | −0.0049 (3) | −0.0137 (3) | 0.0038 (3) |
| O5 | 0.0287 (4) | 0.0598 (6) | 0.0426 (5) | 0.0010 (4) | −0.0082 (3) | 0.0118 (4) |
Geometric parameters (Å, °)
| O1—C2 | 1.2241 (12) | N3—C4 | 1.3716 (12) |
| O2—C4 | 1.2418 (10) | N3—H3 | 0.94 (2) |
| O3—C7 | 1.2056 (13) | C4—C5 | 1.4387 (12) |
| O4—C7 | 1.3040 (13) | C5—C6 | 1.3525 (12) |
| O4—H4 | 0.89 (3) | C5—H5 | 0.879 (19) |
| N1—C6 | 1.3656 (12) | C6—C7 | 1.5012 (12) |
| N1—C2 | 1.3702 (12) | O5—H51 | 0.81 (3) |
| N1—H1 | 0.85 (2) | O5—H52 | 0.82 (3) |
| C2—N3 | 1.3772 (11) | ||
| C7—O4—H4 | 104.3 (16) | N3—C4—C5 | 115.74 (8) |
| C6—N1—C2 | 122.36 (8) | C6—C5—C4 | 118.56 (8) |
| C6—N1—H1 | 126.4 (14) | C6—C5—H5 | 124.1 (11) |
| C2—N1—H1 | 111.2 (14) | C4—C5—H5 | 117.3 (11) |
| O1—C2—N1 | 123.81 (8) | C5—C6—N1 | 122.13 (8) |
| O1—C2—N3 | 121.35 (8) | C5—C6—C7 | 124.03 (9) |
| N1—C2—N3 | 114.83 (8) | N1—C6—C7 | 113.84 (8) |
| C4—N3—C2 | 126.29 (7) | O3—C7—O4 | 125.70 (9) |
| C4—N3—H3 | 120.3 (12) | O3—C7—C6 | 120.28 (10) |
| C2—N3—H3 | 113.3 (12) | O4—C7—C6 | 114.02 (8) |
| O2—C4—N3 | 119.61 (8) | H51—O5—H52 | 110 (2) |
| O2—C4—C5 | 124.64 (9) | ||
| C6—N1—C2—O1 | −178.86 (10) | C4—C5—C6—N1 | −1.20 (15) |
| C6—N1—C2—N3 | 2.20 (15) | C4—C5—C6—C7 | 178.02 (9) |
| O1—C2—N3—C4 | 177.12 (10) | C2—N1—C6—C5 | 0.18 (16) |
| N1—C2—N3—C4 | −3.91 (15) | C2—N1—C6—C7 | −179.11 (9) |
| C2—N3—C4—O2 | −178.17 (10) | C5—C6—C7—O3 | 178.85 (12) |
| C2—N3—C4—C5 | 2.97 (15) | N1—C6—C7—O3 | −1.87 (15) |
| O2—C4—C5—C6 | −179.05 (10) | C5—C6—C7—O4 | −1.13 (15) |
| N3—C4—C5—C6 | −0.26 (14) | N1—C6—C7—O4 | 178.15 (9) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O4—H4···O5 | 0.89 (3) | 1.65 (3) | 2.5231 (11) | 166 (2) |
| N1—H1···O1i | 0.85 (2) | 2.03 (2) | 2.8824 (11) | 175.2 (19) |
| N3—H3···O2ii | 0.94 (2) | 1.87 (2) | 2.8112 (11) | 174.6 (18) |
| O5—H51···O2iii | 0.81 (3) | 2.00 (3) | 2.7786 (12) | 161 (3) |
| O5—H52···O3iv | 0.82 (3) | 1.98 (3) | 2.7787 (12) | 164 (2) |
| C5—H5···O4iii | 0.879 (19) | 2.740 (19) | 3.5922 (13) | 163.7 (15) |
Symmetry codes: (i) −x, −y, −z+2; (ii) −x, −y+1, −z+1; (iii) −x+2, −y, −z+1; (iv) −x+2, −y−1, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: KP2160).
References
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst.32, 115–119.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Brunetti, B., Piacente, V. & Portalone, G. (2000). J. Chem. Eng. Data, 45, 242–246.
- Brunetti, B., Piacente, V. & Portalone, G. (2002). J. Chem. Eng. Data, 47, 17–19.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Motherwell, W. D. S., Shields, G. P. & Allen, F. H. (1999). Acta Cryst. B55, 1044–1056. [DOI] [PubMed]
- Oxford Diffraction (2006). CrysAlis CCD and CrysAlis RED Versions 1.171.32.3. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England.
- Portalone, G., Bencivenni, L., Colapietro, M., Pieretti, A. & Ramondo, F. (1999). Acta Chem. Scand.53, 57–68.
- Portalone, G. & Colapietro, M. (2007). Acta Cryst. C63, o650–o654. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Takusagawa, F. & Shimada, A. (1973). Bull. Chem. Soc. Jpn, 46, 2011–2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680800562X/kp2160sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680800562X/kp2160Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report