

Abstract
Colorless crystals of l-alanine hydrochloride monohydrate, C3H8NO2 +·Cl−·H2O, were obtained from a powder sample that had been left standing in a refrigerator for a few years. The structure displays several intermolecular hydrogen bonds: the hydroxyl O atom is involved in a single hydrogen bond to the chloride anion, while the ammonium group forms one hydrogen bond to the chloride anion and two hydrogen bonds to water molecules. An intermolecular bond between the carbonyl O atom and the ammonium group [2.8459 (15) Å] is also found.
Related literature
For the crystal structures of l-alanine and dl-alanine, see: Simpson & Marsh, (1966 ▶); Dunitz & Ryan, (1966 ▶); Lehmann et al. (1972 ▶); Destro et al. (1988 ▶); Donohue, (1950 ▶); Subha Nandhini et al. (2001 ▶). For the crystal structures of d-alanine hydrochloride and dl-alanine hydrochloride, see: di Blasio et al. (1977 ▶); Trotter, (1962 ▶). For the preparation of the title compound with respect to 17O-labelling, see: Steinschneider et al. (1981 ▶).
Experimental
Crystal data
C3H8NO2 +·Cl−·H2O
M r = 143.57
Orthorhombic,
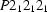
a = 6.1925 (13) Å
b = 9.929 (2) Å
c = 11.759 (3) Å
V = 723.0 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.46 mm−1
T = 150 (2) K
0.45 × 0.40 × 0.35 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker 2001 ▶) T min = 0.819, T max = 0.855
5762 measured reflections
1467 independent reflections
1452 reflections with I > 2σ(I)
R int = 0.022
Refinement
R[F 2 > 2σ(F 2)] = 0.019
wR(F 2) = 0.053
S = 1.11
1467 reflections
84 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.19 e Å−3
Δρmin = −0.20 e Å−3
Absolute structure: Flack (1983 ▶), 533 Friedel pairs
Flack parameter: 0.02 (6)
Data collection: SMART for WNT/2000 (Bruker, 2001 ▶); cell refinement: SAINT-Plus (Bruker, 2001 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808008751/hg2382sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808008751/hg2382Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N—HA⋯O3 | 0.89 | 1.96 | 2.8479 (14) | 174 |
| N—HB⋯Cli | 0.89 | 2.31 | 3.1957 (11) | 171 |
| N—HC⋯O3ii | 0.89 | 1.95 | 2.8380 (15) | 180 |
| O2—H2⋯Cl | 0.82 | 2.23 | 3.0446 (11) | 175 |
| O3—H4⋯Cliii | 0.82 (2) | 2.35 (2) | 3.1432 (12) | 161.0 (17) |
| O3—H5⋯Cliv | 0.78 (2) | 2.38 (2) | 3.1283 (11) | 163.3 (16) |
Symmetry codes: (i) 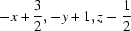 ; (ii)
; (ii) 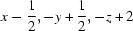 ; (iii)
; (iii) 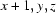 ; (iv)
; (iv) 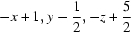 .
.
Acknowledgments
KY appreciates support from the Nanotechnology Support Project of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. SY and TY appreciate support from the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, and MEXT.
supplementary crystallographic information
Comment
L-Alanine is one of the 20 proteinogenic amino acids and has been currently recognized as one of the most abundant amino acids in natural proteins. In general, amino acids very often have polymorphs. The crystal structures of L-alanine (Simpson & Marsh, 1966; Dunitz & Ryan, 1966; Lehmann et al., 1972; Destro et al., 1988), DL-alanine (Donohue, 1950; Subha Nandhini et al., 2001; Trotter, 1962), and D-alanine hydrochloride (di Blasio et al., 1977) have been reported. In the present study, a single-crystal structure determination of L-alanine hydrochloride monohydrate, (I), is reported.
The distances and angles of the present L-alanine molecule are consistent in the typical values reported in the literature of L-alanine molecules (See Table 1 and Figure 1). The atoms N, C2, C3, O1, and O2 are found to be nearly coplanar. The torsion angles of O2—C2—C3—N and O1—C2—C3—N are 174.27 (9) and -6.07 (18), respectively. The angles of O1—C2—O2 and O1—C2—C3, for example, are 125.35 (11) and 123.71 (11)°, respectively. These observations are in reasonable agreement with those of D-alanine hydrochloride reported previously (di Blasio et al., 1977). The torsion angle of O2—C2—C3—C1 was observed to be -64.37 (14)°, which is slightly different from that of L-alanine, the corresponding angle was -76.0° (Lehmann et al., 1972).
The single-crystal diffraction analysis exhibits that the titled compound contains several intermolecular hydrogen bonds. O2 is involved to a single hydrogen bond to a chloride ion with the hydrogen bond distance of 3.0446 (11) Å, while O1 is not involved to hydrogen bonds. Instead, a short contact is observed between O1 and N with the intermolecular bond length of 2.8450 (15) Å. A water molecule donates four hydrogen bonds to two chlorides and two ammonium groups, while a chloride ion accepts four hydrogen bonds from two water molecules, and ammonium and hydroxyl groups (See Table 2 and Figure 2).
It is interesting to compare the present structure with that of D-alanine hydrochloride (di Blasio et al., 1977). In the anhydrous D-alanine hydrochloride crystal, O1 (carbonyl oxygen) exhibits a single hydrogen bond to an ammonium group, and O2 (hydroxyl oxygen) also forms a single hydrogen bond to a chloride ion. The chloride anion, on the other hand, forms three hydrogen bonds, two of which with ammonium groups and one of which with a hydroxyl group. These intermolecular environments are different from the present observations.
Experimental
In the title compound, oxygen-17 isotope enrichments were carried out to the carboxyl group with the aim to perform solid-state 17O NMR experiments. L-alanine hydrochloride was obtained by acid-catalyzed exchange (Steinschneider et al., 1981) with L-alanine and H217O (20% 17O atom, purchased from Taiyo Nippon Sanso Corp., Tokyo, Japan). Colorless crystals of the title compound were obtained from the powder sample after it was left standing in a refrigerator for a few years.
Refinement
All the H atoms except for H4 and H5 were treated as riding atoms with the following distances: for the methyl C—H distance, C—H = 0.96 Å and Uiso(H) = 1.5Ueq(C); for the methyne C—H distance, C—H = 0.98Å and Uiso(H) = 1.2Ueq(C); for the hydroxyl O—H distance, O—H = 0.82Å and Uiso(H) = 1.2Ueq(O); for the ammonium N—H distance, N—H = 0.89Å and Uiso(H) = 1.5Ueq(N). The H4 and H5 atoms were found in difference density Fourier maps, and their positions and isotropic displacement parameters were freely refined.
It might be possible that some degree of racemization occurred since the titled compound had been placed in the refrigerator for a few years. The present diffraction measurements, however, exhibited negligible recemization (the Flack parameter = 0.02 (6)).
It should be noted that "Alert Level B (detecting a pseudo center of symmetry)" was generated by checkCIF/PLATON REPORT during the course of the publication check. The error may come from the fact that the chloride anions are close to a center of symmetry. The present experimental data certainly suggest non-centrosymmetric.
Figures
Fig. 1.

A view of the molecular structure of (I), showing the atom labeling scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

A packing diagram of (I) viewed from a axis. Broken lines indicate the hydrogen bonds.
Crystal data
| C3H8NO2+·Cl–·H2O | F000 = 304 |
| Mr = 143.57 | Dx = 1.319 Mg m−3 |
| Orthorhombic, P212121 | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 902 reflections |
| a = 6.1925 (13) Å | θ = 7.4–53.9º |
| b = 9.929 (2) Å | µ = 0.46 mm−1 |
| c = 11.759 (3) Å | T = 150 (2) K |
| V = 723.0 (3) Å3 | Plate, colourless |
| Z = 4 | 0.45 × 0.40 × 0.35 mm |
Data collection
| Bruker SMART APEX CCD area-detector diffractometer | 1467 independent reflections |
| Radiation source: fine-focus sealed tube | 1452 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.022 |
| T = 293(2) K | θmax = 27.0º |
| ω scans | θmin = 2.7º |
| Absorption correction: multi-scan(SADABS; Bruker 2001) | h = −7→7 |
| Tmin = 0.819, Tmax = 0.855 | k = −11→12 |
| 5762 measured reflections | l = −14→14 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.019 | w = 1/[σ2(Fo2) + (0.0308P)2 + 0.0901P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.053 | (Δ/σ)max = 0.001 |
| S = 1.11 | Δρmax = 0.19 e Å−3 |
| 1467 reflections | Δρmin = −0.20 e Å−3 |
| 84 parameters | Extinction correction: none |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 533 Friedel pairs |
| Secondary atom site location: difference Fourier map | Flack parameter: 0.02 (6) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl | 0.19971 (4) | 0.55991 (3) | 1.25180 (2) | 0.02475 (10) | |
| C1 | 0.8057 (2) | 0.60202 (13) | 0.88317 (11) | 0.0295 (3) | |
| H1A | 0.9301 | 0.6167 | 0.8360 | 0.044* | |
| H1B | 0.7483 | 0.6872 | 0.9073 | 0.044* | |
| H1C | 0.6979 | 0.5540 | 0.8407 | 0.044* | |
| C2 | 0.6780 (2) | 0.49201 (12) | 1.06398 (10) | 0.0249 (2) | |
| N | 0.96092 (17) | 0.38728 (10) | 0.95162 (8) | 0.0230 (2) | |
| HA | 1.0116 | 0.3443 | 1.0124 | 0.034* | |
| HB | 1.0676 | 0.4003 | 0.9021 | 0.034* | |
| HC | 0.8577 | 0.3381 | 0.9193 | 0.034* | |
| O3 | 1.13229 (15) | 0.26883 (10) | 1.15289 (8) | 0.0261 (2) | |
| O1 | 0.60730 (17) | 0.38141 (9) | 1.08180 (8) | 0.0342 (2) | |
| O2 | 0.60094 (18) | 0.60494 (10) | 1.10708 (9) | 0.0374 (2) | |
| H2 | 0.4967 | 0.5880 | 1.1476 | 0.056* | |
| C3 | 0.87031 (18) | 0.51981 (12) | 0.98686 (10) | 0.0228 (2) | |
| H3 | 0.9801 | 0.5694 | 1.0299 | 0.027* | |
| H4 | 1.159 (3) | 0.335 (2) | 1.1930 (16) | 0.047 (5)* | |
| H5 | 1.050 (3) | 0.2271 (18) | 1.1882 (14) | 0.036 (5)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl | 0.02114 (14) | 0.02522 (15) | 0.02790 (15) | 0.00128 (9) | 0.00012 (12) | −0.00516 (12) |
| C1 | 0.0308 (6) | 0.0239 (6) | 0.0337 (6) | 0.0021 (5) | 0.0026 (5) | 0.0073 (5) |
| C2 | 0.0260 (5) | 0.0230 (5) | 0.0256 (5) | 0.0013 (5) | 0.0015 (5) | −0.0018 (5) |
| N | 0.0232 (5) | 0.0200 (5) | 0.0257 (5) | 0.0019 (4) | 0.0028 (4) | 0.0023 (4) |
| O3 | 0.0281 (5) | 0.0235 (4) | 0.0268 (5) | −0.0044 (4) | 0.0023 (4) | −0.0008 (4) |
| O1 | 0.0398 (5) | 0.0238 (4) | 0.0389 (5) | −0.0059 (4) | 0.0160 (4) | −0.0029 (4) |
| O2 | 0.0384 (5) | 0.0244 (4) | 0.0494 (6) | 0.0011 (4) | 0.0183 (5) | −0.0046 (4) |
| C3 | 0.0227 (5) | 0.0183 (5) | 0.0274 (6) | −0.0003 (4) | 0.0011 (5) | −0.0002 (4) |
Geometric parameters (Å, °)
| C1—C3 | 1.5209 (17) | N—HA | 0.8900 |
| C1—H1A | 0.9600 | N—HB | 0.8900 |
| C1—H1B | 0.9600 | N—HC | 0.8900 |
| C1—H1C | 0.9600 | O3—H4 | 0.82 (2) |
| C2—O1 | 1.2006 (16) | O3—H5 | 0.78 (2) |
| C2—O2 | 1.3196 (15) | O2—H2 | 0.8200 |
| C2—C3 | 1.5223 (16) | C3—H3 | 0.9800 |
| N—C3 | 1.4893 (15) | ||
| C3—C1—H1A | 109.5 | C3—N—HC | 109.5 |
| C3—C1—H1B | 109.5 | HA—N—HC | 109.5 |
| H1A—C1—H1B | 109.5 | HB—N—HC | 109.5 |
| C3—C1—H1C | 109.5 | H4—O3—H5 | 104.4 (17) |
| H1A—C1—H1C | 109.5 | C2—O2—H2 | 109.5 |
| H1B—C1—H1C | 109.5 | N—C3—C1 | 110.50 (10) |
| O1—C2—O2 | 125.34 (11) | N—C3—C2 | 107.48 (9) |
| O1—C2—C3 | 123.71 (11) | C1—C3—C2 | 111.65 (11) |
| O2—C2—C3 | 110.95 (10) | N—C3—H3 | 109.1 |
| C3—N—HA | 109.5 | C1—C3—H3 | 109.1 |
| C3—N—HB | 109.5 | C2—C3—H3 | 109.1 |
| HA—N—HB | 109.5 | ||
| O1—C2—C3—N | −6.06 (18) | O1—C2—C3—C1 | 115.28 (14) |
| O2—C2—C3—N | 174.26 (9) | O2—C2—C3—C1 | −64.39 (14) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N—HA···O3 | 0.89 | 1.96 | 2.8479 (14) | 174 |
| N—HB···Cli | 0.89 | 2.31 | 3.1957 (11) | 171 |
| N—HC···O3ii | 0.89 | 1.95 | 2.8380 (15) | 180 |
| O2—H2···Cl | 0.82 | 2.23 | 3.0446 (11) | 175 |
| O3—H4···Cliii | 0.82 (2) | 2.35 (2) | 3.1432 (12) | 161.0 (17) |
| O3—H5···Cliv | 0.78 (2) | 2.38 (2) | 3.1283 (11) | 163.3 (16) |
Symmetry codes: (i) −x+3/2, −y+1, z−1/2; (ii) x−1/2, −y+1/2, −z+2; (iii) x+1, y, z; (iv) −x+1, y−1/2, −z+5/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HG2382).
References
- Bruker (2001). SMART, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Destro, R., Marsh, R. E. & Bianchi, R. (1988). J. Phys. Chem.92, 966–973.
- Blasio, B. di, Pavone, V. & Pedone, C. (1977). Cryst. Struct. Commun.6, 745–748.
- Donohue, J. (1950). J. Am. Chem. Soc.72, 949–953.
- Dunitz, J. D. & Ryan, R. R. (1966). Acta Cryst.21, 617–618.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Lehmann, M. S., Koetzle, T. F. & Hamilton, W. C. (1972). J. Am. Chem. Soc.94, 2657–2660. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Simpson, H. J. & Marsh, R. E. (1966). Acta Cryst.20, 550–555.
- Steinschneider, A., Burgar, M. I., Buku, A. & Fiat, D. (1981). Int. J. Pept. Protein Res.18, 324–333. [DOI] [PubMed]
- Subha Nandhini, M., Krishnakumar, R. V. & Natarajan, S. (2001). Acta Cryst. C57, 614–615. [DOI] [PubMed]
- Trotter, J. (1962). Can. J. Chem.40, 1218–1220.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808008751/hg2382sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808008751/hg2382Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report