

Abstract
The title compound, C10H6ClNO2, has a dihedral angle of 46.46 (5)° between the benzene and maleimide rings. A short intermolecular halogen–oxygen contact is observed, with a Cl⋯O distance of 3.0966 (13) Å. Both CO groups are involved in two C—H⋯O interactions, which gives rise to sheets parallel to (100). In addition, these sheets exhibit a π–π stacking interaction between the benzene and maleimide rings [mean interplanar distance of 3.337 (3) Å].
Related literature
For related literature, see: Etter (1990 ▶); Howell & Zhang (2006 ▶); Metrangolo & Resnati (2001 ▶); Miller et al. (2000 ▶, 2001 ▶); Moreno-Fuquen et al. (2006 ▶); Sureshan et al. (2001 ▶).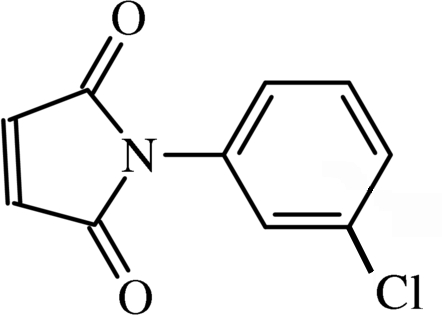
Experimental
Crystal data
C10H6ClNO2
M r = 207.61
Monoclinic,
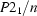
a = 7.3434 (3) Å
b = 11.9458 (5) Å
c = 10.3044 (4) Å
β = 101.121 (2)°
V = 886.96 (6) Å3
Z = 4
Mo Kα radiation
μ = 0.40 mm−1
T = 291 (2) K
0.18 × 0.10 × 0.04 mm
Data collection
Bruker–Nonius KappaCCD diffractometer
Absorption correction: none
4426 measured reflections
2042 independent reflections
1680 reflections with I > 2σ(I)
R int = 0.036
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.101
S = 1.08
2042 reflections
128 parameters
H-atom parameters constrained
Δρmax = 0.25 e Å−3
Δρmin = −0.39 e Å−3
Data collection: DENZO (Otwinowski & Minor, 1997 ▶) and COLLECT (Nonius, 2000 ▶); cell refinement: DENZO and COLLECT; data reduction: DENZO and COLLECT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: PARST95 (Nardelli, 1995 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808011604/fj2114sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011604/fj2114Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C3—H3⋯O2i | 0.93 | 2.53 | 3.455 (2) | 170 |
| C8—H8⋯O2ii | 0.93 | 2.71 | 3.513 (2) | 146 |
| C8—H8⋯O1iii | 0.93 | 2.59 | 3.256 (2) | 129 |
| C2—H2⋯O1iv | 0.93 | 2.72 | 3.308 (2) | 122 |
Symmetry codes: (i) 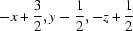 ; (ii)
; (ii) 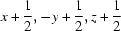 ; (iii)
; (iii) 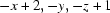 ; (iv)
; (iv) 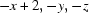 .
.
Acknowledgments
RMF is grateful to the Instituto de Química Física Rocasolano, CSIC, Spain, for the use of the license for the Cambridge Structural Database System (Allen, 2002 ▶). RMF and ZPB acknowledge the Universidad del Valle, Colombia for partial financial support.
supplementary crystallographic information
Comment
Due to the interest created by the N-substituted maleimides in free radical polymerization process upon exposure to light (Howell & Zhang, 2006), the synthesis and study of the crystal structure of N-(m-chlorophenylmaleimide) (I) was undertaken. N-(m-nitrophenylmaleimide) (3NPMI) (Moreno-Fuquen et al., 2006) and N-(o-chlorophenyl) maleimide (2ClPMI) (Miller et al., 2001) systems can be taken as a reference systems to compare with the structural characteristics of (I). Perspective view of (I), showing the atomic numbering scheme, is given in Fig. 1. Photochemical properties of arylmaleimide systems have shown that they depend on the value of the dihedral angle between the benzene and imidic rings (Miller et al., 2000). This angle is 56.2 (1)° and 52.9 (1)° for 3NPMI, 66.10 (4) ° for 2ClPMI, and 46.46 (5)° for (I). The molecules of (I) are linked into sheets by a combination of C—H···O hydrogen bonds (Nardelli, 1995) (Table 1). Indeed, the atoms C3i in the molecule at (3/2 - x, 1/2 + y, 1/2 - z) and C8ii in the molecule at (-1/2 + x, 1/2 - y, -1/2 + z) act as hydrogen-bond donors to maleimidic O2 atom in the molecule at (x, y, z), so generating by 21 screw axis a C(6) chain (Etter, 1990), which is running parallel to [010] direction (Fig. 2, supp. material). Within the asymmetric unit the atom C2 at (x, y, z) acts as hydrogen bond donor to maleimidic O1iii in the molecule at (2 - x, -y, -z), so forming by translation a R22(14) centrosymmetric rings (Etter, 1990); in addition, atom C8 at (x, y, z) acts as a hydrogen bond donor to maleimidic O1ii in the molecule at (2 - x, -y, 1 - z), so generating by translation a R22(8) centrosymmetric rings. Both rings are running along [001] direction (Fig.3, supp. material). In addition, (I) exhibits an aromatic π···π stacking interactions between benzene and maleimide rings with a mean interplanar distance of 3.337 (3) Å. The halogen-oxygen interaction is recognized as a strong driving force in formation of molecular crystals (Sureshan et al., 2001). (I) shows a short Cl···O intermolecular contact, disposed about an inversion centre. The Cl1···O2, shows a distance of 3.0966 (13) Å, [O2 with symmetry 2 - x, 1 - y, -z] and this contact is shorter than the sum of their van der Waalś radii (3.27 Å, Metrangolo & Resnati, 2001). In (I), the angle of the oxygen O2 relative to the C6—Cl bond shows a slight deviation from linearity with a value of 174.31 (6)° and the angle of the chlorine atom relative to the C10—O2 bond shows a value of 136.96 (11)°, suggesting strong halogen bonding. This could also prevent a larger rotation between the planes of (I). The title system does not have enough influence on the processes of polymerization because the dihedral angle between their rings possess a low value with respect to other systems with substituents in the position ortho (Miller et al., 2000).
Experimental
Reagents and solvents for the synthesis were from Aldrich Chemical Co. and they were used without additional purification. Column chromatography was performed using silica gel H60 to purify the intermediates and final products. Thin layer chromatography (TLC) was used to confirm the structure of the individual compounds.
Refinement
The space group P 21/n for (I) was uniquely assigned from the systematic absences. All H-atoms were located from difference maps and then treated as riding atoms [C—H= 0.93Å and Uiso(H)= 1.2Ueq(C)].
Figures
Fig. 1.

An ORTEP-3 (Farrugia, 1997) plot of (I), with the atomic labelling scheme. The shapes of the ellipsoids correspond to 50% probability contours of atomic displacement.
Fig. 2.

Part of the crystal structure of (I) showing the formation of the C(12) and C(13) chains along [010]. Symmetry codes: (i) 3/2 - x, 1/2 + y, 1/2 - z; (ii) -1/2 + x, 1/2 - y, -1/2 + z; (iii) x, 1 + y, z; (iv) -1/2 + x, 1/2 - y, -1/2 + z; (v) 3/2 - x, 3/2 + y, 1/2 - z. For the sake of clarity, the H atoms not involved in the motif shown have been omitted.
Fig. 3.

Part of the crystal structure of (I) showing the formation of a sheet of R22(8) and R22(14) centrosymmetric rings parallel to (100) generated by the C—H···O hydrogen bonds. Symmetry codes: (i) x, y, 1 - z; (ii) -x, -y, -z; (iii) -x, -y, 1 - z; (iv) x, y, 1 + z; (v) -x, -y, 2 - z. For the sake of clarity, the H atoms not involved in the motif have been omitted.
Fig. 4.

The formation of the title compound.
Crystal data
| C10H6ClNO2 | F000 = 424 |
| Mr = 207.61 | Dx = 1.550 Mg m−3 |
| Monoclinic, P21/n | Melting point: 364(1) K |
| Hall symbol: -P 2yn | Mo Kα radiation λ = 0.71073 Å |
| a = 7.3434 (3) Å | Cell parameters from 4426 reflections |
| b = 11.9458 (5) Å | θ = 2.9–27.5º |
| c = 10.3044 (4) Å | µ = 0.40 mm−1 |
| β = 101.121 (2)º | T = 291 (2) K |
| V = 886.96 (6) Å3 | Needle, colorless |
| Z = 4 | 0.18 × 0.10 × 0.04 mm |
Data collection
| Bruker–Nonius KappaCCD diffractometer | 1680 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.036 |
| Monochromator: graphite | θmax = 27.5º |
| φ and ω scans | θmin = 2.9º |
| Absorption correction: none | h = −9→8 |
| 4426 measured reflections | k = −15→15 |
| 2042 independent reflections | l = −13→13 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.038 | w = 1/[σ2(Fo2) + (0.0496P)2 + 0.2501P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.101 | (Δ/σ)max < 0.001 |
| S = 1.09 | Δρmax = 0.25 e Å−3 |
| 2042 reflections | Δρmin = −0.39 e Å−3 |
| 128 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.031 (5) |
| Secondary atom site location: difference Fourier map |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 1.04083 (6) | 0.39876 (4) | −0.16126 (4) | 0.03549 (17) | |
| O1 | 1.08785 (18) | 0.03863 (10) | 0.33200 (12) | 0.0348 (3) | |
| O2 | 0.83189 (18) | 0.38885 (10) | 0.29127 (12) | 0.0348 (3) | |
| N1 | 0.95758 (18) | 0.21271 (11) | 0.27188 (13) | 0.0253 (3) | |
| C1 | 0.8978 (2) | 0.19208 (15) | −0.14130 (16) | 0.0309 (4) | |
| H1 | 0.8853 | 0.1877 | −0.2327 | 0.037* | |
| C2 | 0.8435 (2) | 0.10389 (13) | −0.07035 (17) | 0.0300 (4) | |
| H2 | 0.7944 | 0.0396 | −0.1148 | 0.036* | |
| C3 | 0.8613 (2) | 0.10986 (13) | 0.06635 (16) | 0.0267 (4) | |
| H3 | 0.8224 | 0.0507 | 0.1131 | 0.032* | |
| C4 | 0.9379 (2) | 0.20552 (13) | 0.13225 (15) | 0.0245 (3) | |
| C5 | 0.9937 (2) | 0.29538 (13) | 0.06274 (15) | 0.0267 (4) | |
| H5 | 1.0448 | 0.3594 | 0.1067 | 0.032* | |
| C6 | 0.9710 (2) | 0.28685 (14) | −0.07360 (16) | 0.0285 (4) | |
| C7 | 1.0295 (2) | 0.12740 (14) | 0.36145 (16) | 0.0280 (4) | |
| C8 | 1.0201 (2) | 0.17113 (15) | 0.49571 (16) | 0.0321 (4) | |
| H8 | 1.0594 | 0.1332 | 0.5750 | 0.038* | |
| C9 | 0.9469 (2) | 0.27274 (14) | 0.48355 (16) | 0.0320 (4) | |
| H9 | 0.9263 | 0.3176 | 0.5530 | 0.038* | |
| C10 | 0.9026 (2) | 0.30376 (13) | 0.34085 (16) | 0.0276 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0406 (3) | 0.0386 (3) | 0.0306 (3) | 0.00442 (17) | 0.01531 (18) | 0.01000 (17) |
| O1 | 0.0433 (7) | 0.0275 (6) | 0.0336 (7) | 0.0019 (5) | 0.0076 (5) | 0.0034 (5) |
| O2 | 0.0438 (7) | 0.0307 (7) | 0.0318 (7) | 0.0066 (5) | 0.0122 (5) | 0.0004 (5) |
| N1 | 0.0308 (7) | 0.0245 (7) | 0.0215 (7) | −0.0005 (5) | 0.0071 (5) | 0.0015 (5) |
| C1 | 0.0321 (8) | 0.0380 (9) | 0.0236 (8) | 0.0098 (7) | 0.0074 (6) | −0.0012 (7) |
| C2 | 0.0310 (8) | 0.0294 (8) | 0.0290 (8) | 0.0061 (6) | 0.0047 (7) | −0.0048 (6) |
| C3 | 0.0277 (8) | 0.0254 (8) | 0.0270 (8) | 0.0030 (6) | 0.0056 (6) | 0.0007 (6) |
| C4 | 0.0245 (7) | 0.0270 (8) | 0.0228 (7) | 0.0039 (6) | 0.0063 (6) | 0.0005 (6) |
| C5 | 0.0284 (8) | 0.0280 (8) | 0.0251 (8) | 0.0015 (6) | 0.0083 (6) | −0.0001 (6) |
| C6 | 0.0286 (8) | 0.0314 (8) | 0.0277 (8) | 0.0064 (6) | 0.0110 (6) | 0.0043 (6) |
| C7 | 0.0295 (8) | 0.0278 (8) | 0.0265 (8) | −0.0057 (6) | 0.0050 (6) | 0.0039 (6) |
| C8 | 0.0376 (9) | 0.0355 (9) | 0.0237 (8) | −0.0080 (7) | 0.0073 (7) | 0.0024 (7) |
| C9 | 0.0389 (9) | 0.0351 (9) | 0.0240 (8) | −0.0076 (7) | 0.0109 (7) | −0.0021 (7) |
| C10 | 0.0290 (8) | 0.0285 (8) | 0.0270 (8) | −0.0032 (6) | 0.0093 (6) | −0.0012 (6) |
Geometric parameters (Å, °)
| Cl1—C6 | 1.7450 (17) | C3—C4 | 1.392 (2) |
| O1—C7 | 1.204 (2) | C3—H3 | 0.9300 |
| O2—C10 | 1.209 (2) | C4—C5 | 1.395 (2) |
| N1—C10 | 1.401 (2) | C5—C6 | 1.386 (2) |
| N1—C7 | 1.408 (2) | C5—H5 | 0.9300 |
| N1—C4 | 1.421 (2) | C7—C8 | 1.493 (2) |
| C1—C6 | 1.383 (2) | C8—C9 | 1.324 (3) |
| C1—C2 | 1.384 (2) | C8—H8 | 0.9300 |
| C1—H1 | 0.9300 | C9—C10 | 1.490 (2) |
| C2—C3 | 1.391 (2) | C9—H9 | 0.9300 |
| C2—H2 | 0.9300 | ||
| C10—N1—C7 | 109.76 (13) | C4—C5—H5 | 120.9 |
| C10—N1—C4 | 125.30 (13) | C1—C6—C5 | 122.04 (15) |
| C7—N1—C4 | 124.90 (13) | C1—C6—Cl1 | 119.38 (13) |
| C6—C1—C2 | 118.70 (15) | C5—C6—Cl1 | 118.57 (13) |
| C6—C1—H1 | 120.6 | O1—C7—N1 | 125.39 (15) |
| C2—C1—H1 | 120.6 | O1—C7—C8 | 128.61 (16) |
| C1—C2—C3 | 121.03 (15) | N1—C7—C8 | 106.00 (14) |
| C1—C2—H2 | 119.5 | C9—C8—C7 | 108.89 (15) |
| C3—C2—H2 | 119.5 | C9—C8—H8 | 125.6 |
| C2—C3—C4 | 119.09 (15) | C7—C8—H8 | 125.6 |
| C2—C3—H3 | 120.5 | C8—C9—C10 | 109.20 (15) |
| C4—C3—H3 | 120.5 | C8—C9—H9 | 125.4 |
| C3—C4—C5 | 120.84 (15) | C10—C9—H9 | 125.4 |
| C3—C4—N1 | 119.76 (14) | O2—C10—N1 | 125.51 (15) |
| C5—C4—N1 | 119.40 (14) | O2—C10—C9 | 128.34 (15) |
| C6—C5—C4 | 118.28 (15) | N1—C10—C9 | 106.14 (14) |
| C6—C5—H5 | 120.9 | ||
| C6—C1—C2—C3 | 0.2 (2) | C10—N1—C7—O1 | 179.89 (16) |
| C1—C2—C3—C4 | −1.1 (2) | C4—N1—C7—O1 | 1.9 (3) |
| C2—C3—C4—C5 | 1.1 (2) | C10—N1—C7—C8 | −0.97 (17) |
| C2—C3—C4—N1 | −179.78 (14) | C4—N1—C7—C8 | −178.92 (14) |
| C10—N1—C4—C3 | −131.50 (16) | O1—C7—C8—C9 | 179.73 (17) |
| C7—N1—C4—C3 | 46.1 (2) | N1—C7—C8—C9 | 0.62 (18) |
| C10—N1—C4—C5 | 47.7 (2) | C7—C8—C9—C10 | −0.05 (19) |
| C7—N1—C4—C5 | −134.69 (16) | C7—N1—C10—O2 | −178.35 (16) |
| C3—C4—C5—C6 | −0.1 (2) | C4—N1—C10—O2 | −0.4 (3) |
| N1—C4—C5—C6 | −179.23 (14) | C7—N1—C10—C9 | 0.94 (17) |
| C2—C1—C6—C5 | 0.9 (2) | C4—N1—C10—C9 | 178.88 (14) |
| C2—C1—C6—Cl1 | 179.82 (12) | C8—C9—C10—O2 | 178.72 (17) |
| C4—C5—C6—C1 | −0.9 (2) | C8—C9—C10—N1 | −0.54 (19) |
| C4—C5—C6—Cl1 | −179.88 (11) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O2i | 0.93 | 2.53 | 3.455 (2) | 170 |
| C8—H8···O2ii | 0.93 | 2.71 | 3.513 (2) | 146 |
| C8—H8···O1iii | 0.93 | 2.59 | 3.256 (2) | 129 |
| C2—H2···O1iv | 0.93 | 2.72 | 3.308 (2) | 122 |
Symmetry codes: (i) −x+3/2, y−1/2, −z+1/2; (ii) x+1/2, −y+1/2, z+1/2; (iii) −x+2, −y, −z+1; (iv) −x+2, −y, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2114).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Etter, M. (1990). Acc. Chem. Res.23, 120–126.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Howell, B. & Zhang, J. (2006). J. Therm. Anal. Calorim.83, 83–86.
- Metrangolo, P. & Resnati, G. (2001). Chem. Eur. J.7, 2511–2519. [DOI] [PubMed]
- Miller, C. W., Hoyle, C. E., Valente, E. J., Zobkowski, J. D. & Jönsson, E. S. (2000). J. Chem. Crystallogr.30, 9, 563–571.
- Miller, C. W., Jönsson, E. S., Hoyle, C. E., Viswanathan, K. & Valente, E. J. (2001). J. Phys. Chem. B, 105, 2707–2717.
- Moreno-Fuquen, R., Valencia, H., Pardo, Z. D., D’Vries, R. & Kennedy, A. R. (2006). Acta Cryst. E62, o2734–o2735.
- Nardelli, M. (1995). J. Appl. Cryst.28, 659.
- Nonius (2000). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr and R. M. Sweet, pp. 307–326. New York: Academic Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sureshan, K. M., Gonnade, R. G., Shashidhar, M. S., Puranik, V. G. & Bhadbhade, M. M. (2001). Chem. Commun. pp. 881–882.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808011604/fj2114sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011604/fj2114Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report