


Abstract
There is still a lack of information on the specific characteristics of DNA-binding proteins from hyperthermophiles. Here we report on the product of the gene orf56 from plasmid pRN1 of the acidophilic and thermophilic archaeon Sulfolobus islandicus. orf56 has not been characterised yet but low sequence similarily to several eubacterial plasmid-encoded genes suggests that this 6.5 kDa protein is a sequence-specific DNA-binding protein. The DNA-binding properties of ORF56, expressed in Escherichia coli, have been investigated by EMSA experiments and by fluorescence anisotropy measurements. Recombinant ORF56 binds to double-stranded DNA, specifically to an inverted repeat located within the promoter of orf56. Binding to this site could down-regulate transcription of the orf56 gene and also of the overlapping orf904 gene, encoding the putative initiator protein of plasmid replication. By gel filtration and chemical crosslinking we have shown that ORF56 is a dimeric protein. Stoichiometric fluorescence anisotropy titrations further indicate that ORF56 binds as a tetramer to the inverted repeat of its target binding site. CD spectroscopy points to a significant increase in ordered secondary structure of ORF56 upon binding DNA. ORF56 binds without apparent cooperativity to its target DNA with a dissociation constant in the nanomolar range. Quantitative analysis of binding isotherms performed at various salt concentrations and at different temperatures indicates that approximately seven ions are released upon complex formation and that complex formation is accompanied by a change in heat capacity of –6.2 kJ/mol. Furthermore, recombinant ORF56 proved to be highly thermostable and is able to bind DNA up to 85°C.
INTRODUCTION
Sulfolobus ssp., an acidophilic and thermophilic member of the Crenarchaeota, is an attractive model organism of the archaea, the third kingdom of life, because it is easy to cultivate and methods for its genetic manipulation have been developed (1,2).
The plasmid pRN1, first isolated by W.Zillig (3) from Sulfolobus islandicus, has been fully sequenced (4) (accession no. U36383). pRN1 belongs to the archael pRN plasmid family (5), which comprises the plasmids pRN1, pRN2 (6), pSSVx (7), pHEN7 (5) and pDL10 (8). All except pDL10 were isolated from S.islandicus strains; pDL10 was isolated from the crenarcheote Acidianus ambivalens. These five circular plasmids have three open reading frames (ORFs) in common (5,6). Only one of the three ORF groups has sequence similiarity to proteins with known function. Specifically, orf56 from pRN1 is similiar to several eubacterial rolling circle plasmid-encoded DNA-binding proteins with proven or suggested function as copy control proteins (Fig. 1). The genes of these copy control proteins are located upstream of the genes for initiator proteins for plasmid replication and are transcribed from a promoter upstream of the copy control gene. As shown for plasmids pLS1 and pE194, the copy control proteins bind to their own promoter and thereby down-regulate their own synthesis and synthesis of the initiator protein of plasmid replication. In addition to this feedback control, synthesis of the replicative initiator protein is regulated by counter-transcript RNA, preventing translation of the initiator protein (9,10).
Figure 1.

Alignment of ORF56 from pRN1 with copy control proteins (CopG) from other rolling circle plasmids. ORFC from pWVO1 (accession no. JQ1198), CopA from ppsc22 (accession no. X95843), Cop protein from pSBO2 (accession no. AB021465), CopG (formerly RepA) from pLS1 (accession no. A25599), ORF52 from pRN2 (accession no. U93082), Cop-6 from pE194 (accession no. M59209) and ORF56 from pRN1 (accession no. U36383). DNA binding has been shown for CopG from pLS1 (26) and Cop-6 (27). The GOR IV secondary structure prediction (c, coil; e, β-strand; h, α-helix) is given below the alignment.
Similarly to eubacterial rolling circle plasmids, the gene of the putative copy control protein orf56 overlaps with the gene orf904, which does not seem to have a promoter of its own (4). The gene orf904 codes for a large protein with a helicase domain and could function as the initiator protein of plasmid replication. There is evidence that plasmid pDL10 replicates via a single-stranded intermediate, which suggests rolling circle replication for the plasmid family pRN (8).
Due to its small size plasmid pRN1 is an attractive backbone for constructing a high copy Escherichia coli–Sulfolobus shuttle vector. A shuttle vector would facilitate genetic studies with Sulfolobus solfataricus, whose genome sequence is being completed. However, so far none of the gene products of this plasmid family have been characterised.
The sequence-specific recognition of DNA is of the utmost importance for regulation of gene-specific transcription. The thermodynamics of sequence-specific binding has been intensively studied. For several proteins, e.g. EcoRI endonuclease and the lac repressor, it has been shown that the binding reaction is entropic at lower temperatures and enthalpic at higher temperatures. For the mesophilic proteins studied binding is optimal at the physiological temperature (11).
In this communication we report on the heterologous expression of ORF56, its purification and characterisation of its DNA-binding activity. We show that ORF56 binds preferentially within its promoter region.
MATERIALS AND METHODS
Oligodeoxynucleotides
All oligodeoxynucleotides were purchased from Eurogentec (Seraing, Belgium), Genosys (Cambridge, UK) and Interactiva (Ulm, Germany). Oligodeoxynucleotides used for DNA-binding assays were quality checked by radioactive labelling with T4 polynucleotide kinase followed by denaturing polyacrylamide gel electrophoresis.
Double-stranded DNA for the fluorescence measurements was obtained by annealing the complementary oligonucleotides at 100 µM in 1× NEB Buffer 4 (New England Biolabs, Beverly, MA) in a thermocycler with the following temperature profile: 2 min at 95°C, 6 min at 65°C, followed by cooling to 25°C at 1.2°C/min. Complete hybridisation was checked by post-labelling with T4 polynucleotide kinase followed by native polyacrylamide gel electrophoresis
Cloning of orf56 and overexpression of ORF56
Escherichia coli XL1-Blue was used for cloning. The orf56 gene was amplified from plasmid pUC18-pRN1 (a gift from David Faguy, Dalhousie University, Canada) by PCR with the forward primer 5′-GGAATTCCATATGGCCATGGGTAGACCATAC and the reverse primer 5′-GATAAAGAAAAGAAGTAACTCGAGGGATCCCG. The PCR product was cut with NcoI and XhoI (New England Biolabs) and ligated into the expression vector pET28c (Novagen) cut with the same enzymes. The identity of the insert of the resulting plasmid (pET28-orf56) was confirmed by DNA sequencing. The final construct was transformed into E.coli BL21 (DE3, pLysS) cells which were used for overexpression. Aliquots of 4 ml of an overnight culture were inoculated into 1 l of LB medium supplemented with 50 µg/ml kanamycin and 34 µg/ml chloramphenicol and grown at 37°C. After 3 h 1 mM IPTG was added and the culture was fermented for a further 3 h. Then the cells were pelleted and kept frozen at –70°C until use.
Purification of ORF56
The frozen cells were resuspended in 20 ml of lysis buffer (50 mM sodium phosphate, 1 mM EDTA, pH 8.0). Lysozyme (100 µg/ml) and Triton X-100 (0.1%) were added and the cells were incubated for 15 min at 4°C. After sonication the lysate was clarified by centrifugation for 30 min at 4°C and 40 000 g. The supernatant was incubated for 15 min at 70°C and the denatured host proteins were removed by a second centrifugation step (30 min, 4°C, 40 000 g). The NaCl concentration was raised to 100 mM NaCl and the pH was adjusted to pH 8.0 before the extract was loaded onto a 2.6 × 4 cm EMD-SO3– column (Merck, Darmstadt, Germany) equilibrated with 50 mM potassium phosphate, 200 mM NaCl, pH 8.0. The column was developed with a linear gradient of 200–800 mM NaCl. Pure ORF56 eluted at 400 mM NaCl. The protein was frozen immediately at –70°C or dialysed against 25 mM potassium phosphate, 30% (v/v) glycerol, pH 8.0, for storage at –70°C. Total yield from 1 l of expression culture was 35 mg protein as determined by absorption at 280 nm using the theoretical extinction coefficient ɛ = 9590 M–1cm–1.
Gel filtration
Gel filtration was carried out on a Toso Haas TSK G3000SW (60 × 0.75 cm) column. Aliquots of 20 µl of ORF56 (500, 100, 20 and 0.4 µM monomer concentration) were loaded at 0.5 ml/min onto a column equilibrated with 100 mM sodium sulphate, 100 mM sodium phosphate, pH 7.2. The molecular mass M was calculated using linear regression between logM and Kav = (Ve – V0)/(Vt – V0). The coefficient of regression r2 was at least 0.99.
Circular dichroism spectra
The CD spectra were recorded with a thermostated Jasco J600 CD spectropolarimeter. Wavelength scans were performed at 20°C in 0.2 nm steps from 194 to 260 nm in 25 mM sodium phosphate, 100 mM KCl, pH 7.6, 0.5 mM polyoxyethylene 8-decylether (Sigma). The protein concentration was 10 µM monomer and the DNA concentration was 3.3 µM. The spectra were corrected for the spectra of the buffer and the DNA. For the titrations the DNA concentration was varied from 0 to 4.2 µM. The CD signal at 213 nm (2 nm slit) was monitored for 2 min at each titration point and corrected for the small background signal of the DNA alone. Temperature scans were done in 0.5°C steps from 20 to 100°C. Protein concentration was 2.5 and 25 µM for the temperature scan. All measurements were done in cuvettes with 1 mm path length.
Dimethylsuberimidate crosslinks
ORF56 (20 pmol) was incubated with and without 120 pmol double-stranded binding site DNA (wt) and control DNA (ct) in the presence of 10 mM DMS in 0.2 M triethanolamine, pH 8.0, for 1 h at room temperature in a reaction volume of 20 µl. Aliquots of the reactions were separated on a 16% polyacrylamide peptide gel according to Schagger and von Jagow (12).
Electrophoretic mobility shift assays (EMSA)
The 167 bp PCR fragment (promoter region, Fig. 5) was labelled uniformly by including [α-35S]dATP (Hartmann, Braunschweig, Germany) in the PCR. The PCR fragment was digested with the restriction endonuclease MseI and the digestion products were used as probes for the EMSA. The binding reaction was performed with 3 µM ORF56 in assay buffer (20 mM Tris–HCl, 100 mM KCl, 1 mM EDTA, 1 mM DTT, pH 7.6) for 10 min at 65°C. A sample (5–10 µl) was loaded with 1–2 µl of loading buffer (0.1% xylene cyanol, 0.1% bromphenol blue, 0.1 mM EDTA in 75% glycerol) onto a 6% polyacrylamide gel in TAE (40 mM Tris–acetate, 1 mM EDTA). The gel was dried and analysed with an Instant Imager (Canberra Packard). The image intensity is linear with respect to the signal.
Figure 5.

The promoter region of the orf56 gene. Thin horizontal arrows indicate primers used for amplifying the promoter DNA. The primers for amplifying the promoter region were 5′-TCTCTACTTTCCTAACTCTCTG and 5′-GTATGGTCTACCCATTTG. The sequence immediately upstream of the start codon is shown in detail. Thick arrows indicate the two imperfect inverted repeats within the 47 bp MseI fragment. The putative BRE, TATA box and box B of the promoter are also indicated. The boxes were assigned according to rules formulated by Reiter et al. (24), Qureshi and Jackson (34) and Bell et al. (25).
For EMSA with synthetic DNA, 10 pmol oligodeoxynucleotide (see Table 1) was 5′-labelled with 1 pmol [γ-32P]ATP (Hartmann) using T4 polynucleotide kinase (Fermentas). The labelled oligodeoxynucleotide was hybridised with a 20% excess of the complementary oligodeoxynucleotide. The synthetic double-stranded DNA (5 nM) was used as probe as described above.
Table 1. Synthetic DNA substrates used to evaluate the DNA-binding activity of ORF56.
 |
|
Alignment of the synthetic DNA substrates used in this study. wt is derived from the longer inverted repeat within the promoter region of gene orf56 (Fig. 5). wt+CGC has three additional bases at both termini in order to increase the melting temperature of the corresponding duplex DNA. rep is the half-site sequence of the wt DNA. The synthetic substrate inv is derived from the shorter inverted repeat within the promoter region. ct is a control DNA used in the study. The arrows indicate repeat structures. Only one strand is shown. Double-stranded DNAs were used in all experiments.
Fluorescence anisotropy measurements
Fluorescence anisotropy measurements were performed on a Perkin Elmer LS-50B fluorescence spectrometer equipped with a thermojacket and a stirrer. As substrate we used wt DNA (Table 1) carrying a fluorescein via a hexyl linker at one 3′-end. Typically, 100 nM ORF56 was added to a solution of 2 nM fluorescein-labelled DNA. The ORF56 concentration was then serially decreased down to 0.2 nM final monomer concentration by replacing aliquots of the ORF56/DNA solution in the cuvette with buffer containing 2 nM DNA. Titration was carried out in 20 mM Tris–HCl, pH 7.5, 0.01% Tween 20, 100 mM KCl (standard buffer) in a 1 ml cuvette equipped with a stirrer and thermostated at 25°C. The cuvette was silanised with 2% (v/v) dichlorodimethylsilane in trichloroethane (Merck, Darmstadt, Germany). The sample was excited at 495 nm using a 15 nm slit and a vertical polarising filter. The vertical and horizontal emission was monitored at 528 nm with a slit of 20 nm and a cut-off filter of 515 nm. The grating factor was calculated with fluorescein as the depolarising sample. At each titration point anisotropy was measured at least four times with a 10 s integration time.
The experimental data was fitted to the following model.
A4 + B ⇔ A4B Kd 1
![]()
![]() 2
2
[A4B] represents the concentration of the DNA–protein complex and [A0] and [B0] the concentration of total protein monomer and the total DNA concentration.
Since the fluorescence intensity of the fluorescein-labelled DNA did not change upon ORF56 binding, the fluorescence anisotropy is given by:
![]() 3
3
where r is the observed anisotropy and rB and rA4B are the anisotropy of the free and bound DNA.
The data sets were least square fitted using the Levenberg–Marquardt algorithm.
Stoichiometric titrations were carried out with the following modifications. The DNA concentration was raised to 50 nM and the excitation and emission slits were set to 10 and 12 nm, respectively.
The salt dependence was studied in 10 mM Tris–HCl, pH 7.5, 0.01% Tween 20 with the KCl concentration varied between 125 and 300 mM. For each salt concentration 10–13 ORF56 concentrations spanning about two orders of magnitude were titrated.
To study the temperature dependence, 5 nM wt DNA was titrated with ORF56 ranging from 2.77 to 200 nM (13 data points). The assay buffer for this experiment was 20 mM potassium cacodylate, pH 7.5, 0.01% Tween 20, 150 mM KCl. The isotherms from 17 to 47°C were globally fitted assuming a linear temperature dependence of the anisotropy of the free DNA and of the complex. A linear relationship was found for free probe in the temperature range investigated and it was reasonable to use a linear relationship for the complex as well. The binding isotherm at 57°C deviated strongly and was fitted separately. The global fit yielded the temperature-dependent Kd, which was then analysed according to (13):
lnKd = –ΔCp/R/T*{(TH/T) – 1 – ln(TS/T)} 4
where ΔCp is the temperature-independent heat capacity change for the association reaction and TH and TS are the temperatures at which ΔH and ΔS, respectively, are 0.
For the competition experiments 2 nM fluorescently labelled wt DNA was allowed to bind to 20 nM ORF56. Then increasing amounts of unlabelled DNA were added and the decrease in anisotropy was recorded. The resulting isotherms were fitted with the computer program Dynafit (14) considering two equilibria. ORF56 binds to the fluorescently labelled wt DNA or to the competitor DNA with dissociation constant Kd or Kc, respectively. For genomic DNA we assumed that the number of non-specific binding sites equals the number of base pairs. All binding sites have the same microscopic dissociation constant in this calculation. We also assume that the sites overlap, which is correct given the large excess of binding sites over the protein concentration. For the competition experiment with the single repeat DNA the calculation is based on the dimer concentration.
Thermal denaturation of DNA
Double-stranded DNA at a concentration of 1 µM was incubated in 20 mM Tris–HCl, 100 mM KCl, pH 7.6, with and without 4 µM ORF56 in a 1 ml cuvette. For this experiment a modified wt DNA was used which had extra GCG bases at both termini in order to increase its melting temperature. The absorbance at 260 nm was monitored while the cuvette was heated with a Peltier element from 50 to 99°C in steps of 0.5°C, allowing 30 s for equilibration. The melting temperature Tm was calculated by fitting the temperature profile assuming a constant ΔCp and that the slopes of the single- and double-stranded baselines (mS and mD) do not differ between the sample with and without protein (15).
![]() ×
×
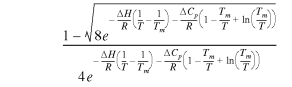 5
5
where A is the corrected and normalised absorption at 260 nm, ΔH the renaturation enthalpy at Tm and yD0 and yS0 the extrapolated absorptions of the single- and double-stranded DNA at 0 K.
RESULTS
Expression and purification
The gene orf56 was cloned into the E.coli expression vector pET28c (Novagen) under control of the T7 promoter. Upon induction BL21 (DE3, pLysS) E.coli cells expressed the recombinant protein to ∼10% of total protein. The thermophilic protein was purified by a heat denaturing step, removing the bulk of host proteins, and by cation exchange chromatography. Pure protein eluted at 400 mM NaCl from the EMD-SO3– column (Fig. 2). The calculated molecular mass of 6655 Da agrees well with its retention in Tricine–SDS gels. From sequencing of the purified protein the N-terminus of the protein was determined to be GRPYK, indicating removal of the initiator methionine by the E.coli host cells.
Figure 2.
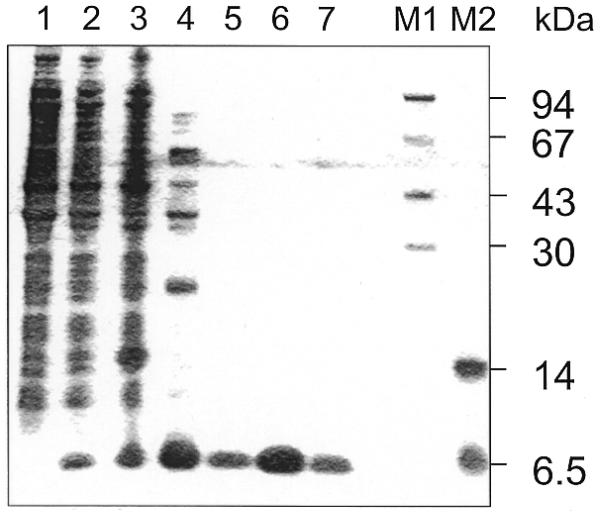
Purification of recombinant ORF56. Protein from various steps of the purification protocol was analyzed on a 16% SDS–Tris–Tricine polyacrylamide gel (12) stained with Coomassie Blue. Lane 1, whole cell extract of an uninduced control culture; lane 2, whole cell extract of an induced E.coli (pET28-orf56) culture; lane 3, crude extract (centrifuged whole cell extract) of an induced culture; lane 4, heat step supernatant; lanes 5–7, pooled peak fractions of purified ORF56; lanes M1 and M2, molecular mass markers.
Secondary structure and dimerisation
The secondary structure of ORF56 was evaluated by UV CD spectroscopy (Fig. 3A). The negative ellipticity in the wavelength region 200–230 nm with molar ellipticities down to –16 000 deg cm2/dmol indicates a high α-helical content of ORF56. Deconvolution of the spectrum with a neural network (16) results in 52% α-helix and 9% β-strand conformation. This analysis agrees well with the secondary structure prediction according to GOR IV (17). This algorithm predicts a short β-strand which is followed by two longer helices (Fig. 1). The N-terminus as well as the C-terminus are predicted to have an unordered secondary structure.
Figure 3.


UV CD spectra of ORF56. (A) CD spectra of 10 µM ORF56 free (continuous line) and complexed with 3.3 µM wt DNA (broken line) corrected for the background of DNA alone in standard buffer. (B) Decrease in the CD signal at 213 nm as a function of added wt DNA. The titration is stoichiometric (10 µM ORF56).
The binding of ORF56 to binding site DNA (see below) has a significant effect on its secondary structure. The CD spectrum of the protein–DNA complex shows a decrease in optical ellipticity in the spectral region 210–230 nm as compared to the free protein (Fig. 3A). The decrease is indicative of additional formation of α-helical regions or of type II β-turns. The change in ellipticity increases with the amount of added DNA up to stoichiometric equivalence (Fig. 3B). Possibly the predicted coiled regions at the termini become structured upon binding to the DNA. We did not observe any significant changes in the CD spectrum above 240 nm, which would indicate change of the B-DNA conformation to the A or Z conformation (18).
The 6.5 kDa protein ORF56 elutes from a gel filtration column with an apparent molecular mass of 17 kDa (data not shown). We ascribe this observation to a non-ideal elution behaviour of the 13 kDa dimeric ORF56. Additional evidence that ORF56 is dimeric in solution comes from chemical crosslinking with the bifunctional amino-reactive reagent dimethylsuberimidate (DMS). When ORF56 is incubated with DMS and the reaction products are separated on a denaturing peptide gel, a protein of ∼14 kDa is visible (Fig. 4, lane 2). This protein has the expected molecular mass of a crosslink between two ORF56 monomers.
Figure 4.
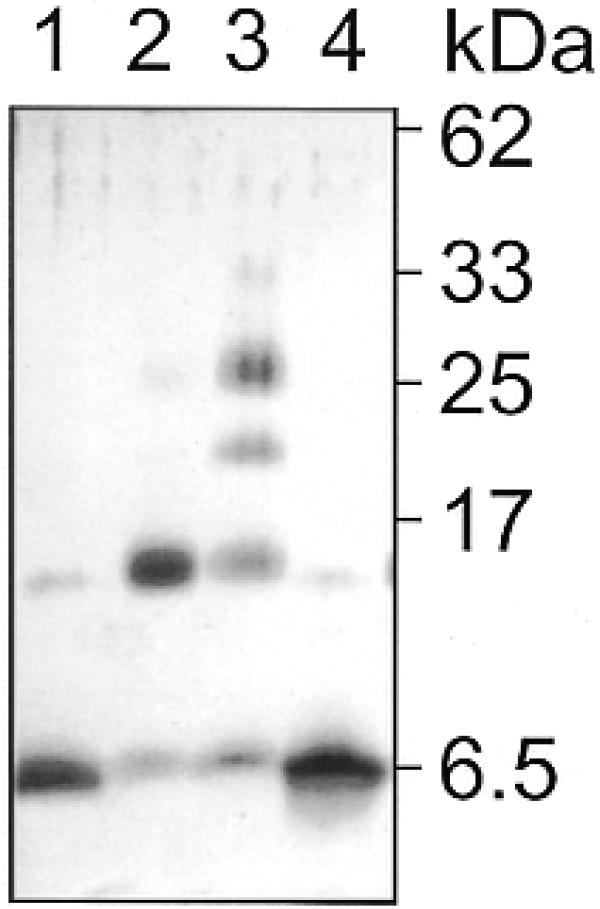
Chemical crosslinking of ORF56 with DMS. ORF56 was incubated with and without crosslinker and analyzed on a denaturing peptide gel. Lane 1, no DMS; lane 2, 10 mM DMS added. The crosslinking reaction was repeated in the presence of binding site DNA (wt): lane 3, 10 mM DMS; lane 4, no DMS.
When chemical crosslinking is done in the presence of binding site DNA two additional proteins with apparent molecular masses of 20 and 27 kDa are produced (Fig. 4, lane 3). These proteins are likely to be chemical crosslinks between three and four ORF56 polypeptides. The gel filtration and the DMS crosslink results suggest that ORF56 is dimeric in solution and that ORF56 could bind as a tetramer to its binding site DNA. Additional evidence that ORF56 is tetrameric on the DNA comes from stoichiometric fluorescence titrations (see below).
The secondary structure prediction (β-sheet followed by helix–turn–helix), the CD spectroscopy, the gel filtration and the chemical crosslinking data suggest that ORF56 might adopt a fold similar to the CopG protein. The crystal structure of CopG from pLS1, both unliganded and complexed with DNA, has been solved (19). The structures show a short β-sheet followed by a helix–turn–helix motif. In solution CopG is dimeric. The N-terminal amino acids of the two monomers form a joint antiparallel β-sheet, which binds to the major groove of the DNA. A tetramer assembles on the inverted repeat of the target DNA, with each β-sheet recognising one repeat.
ORF56 binds specifically to its own promoter DNA
First we studied the DNA-binding activity of ORF56 by gel retardation assays with DNA fragments upstream of the orf56 gene. An internally radioactively labelled 167 bp long PCR fragment (Fig. 5) was digested with MseI, yielding three fragments of 61, 59 and 47 bp. Of these fragments, only the 47 bp DNA fragment was shifted by addition of ORF56 in the presence of 40 ng/µl competitor DNA, indicating specific binding to this fragment only (Fig. 6A). The 47 bp fragment contains the initiator methionine codon and two imperfect inverted repeats (Fig. 5). To further narrow down the binding site of ORF56, oligonucleotides were derived from the 47 bp restriction fragment and used as probes in EMSA (Table 1). Both inverted repeats (inv and wt) were assayed separately, but only the probe containing the longer inverted repeat (wt) was bound (Fig. 6B). Furthermore, we did not observe any binding to a single repeat (rep) and to the single-stranded inverted repeat wt (data not shown).
Figure 6.
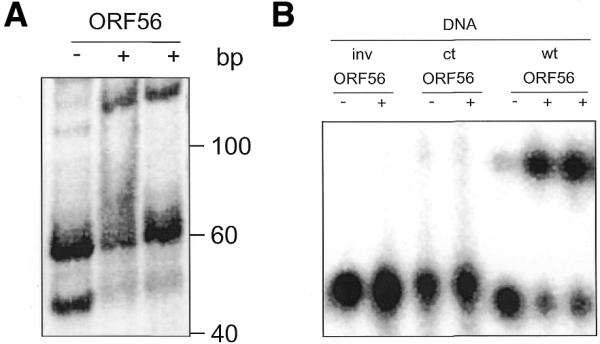
Specific binding of ORF56 analysed by EMSA. (A) A MseI restriction digest of the promoter PCR fragment (61, 59 and 47 bp) was probed with 3 µM ORF56. The 47 bp fragment is bound by ORF56 even in the presence of 40 ng/µl competitor DNA (right-most lane). (B) Binding of ORF56 (5 µM) to synthetic 32P-labelled duplex DNA substrates. inv, short inverted repeat; ct, negative control DNA with no repeat structure; wt, longer inverted repeat. The wt DNA was also probed in the presence of 200-fold excess of poly[d(I·C)·d(I·C)] (right-most lane). See Table 1 and Figure 5 for the sequences and the positions of the synthetic DNA substrates.
In addition to the EMSA experiments we tried to determine the binding site of ORF56 through DNase I footprinting. Footprinting experiments with DNase I did not produce a clear footprint.
Strength of DNA binding
The EMSA experiments only give a crude measure of the binding affinity and the observed binding was rather weak, with dissociation constants in the micromolar range. To characterise the binding equilibria in more detail we applied fluorescence anisotropy measurements (20). For these assays we used duplex wt DNA which was labelled with fluorescein at one 3′-end. The fluorescence anisotropy of the DNA increases upon complexation with protein because rotational diffusion of the complex is reduced compared to unbound DNA.
Figure 7A shows titration of 2 nM fluorescently labelled wt DNA with ORF56. The data could be very well fitted assuming a simple one-site model. The anisotropy of the free probe was 0.099 and the anisotropy of the DNA–protein complex was 0.159. Half-maximal binding was at ∼8 nM ORF56 monomers. Using a competition experiment with unlabelled wt DNA we could show that both DNAs, the unlabelled and the fluorescently labelled, were bound equally well by ORF56 (Fig. 7B). Furthermore, the fluorescence intensity and the emission spectra of bound and complexed probe were virtually identical. Therefore, we can safely assume that the fluorescein moiety attached at the 3′-end of one strand does not interfere with binding of ORF56 to this substrate.
Figure 7.



Fluorescence anisotropy titration of fluorescently labelled wt DNA with ORF56. (A) Titration of 2 nM fluorescein-labelled wt DNA. The experimental points and a fit based on the A4B model are given. The error bars are the standard errors of each titration point calculated from four to six measurements. In this fit the anisotropy of the unbound DNA is 0.099 and 0.159 for the bound DNA. The dissociation constant based on the tetramer concentration is 1.1 nM. (B) Competition experiment with 2 nM fluorescein-labelled wt DNA/20 nM ORF56 and increasing amounts of unlabelled wt DNA. The fit of the data yields a dissociation constant for the unlabelled DNA of 0.88 nM. (C) Stoichiometric titration of 50 nM fluorescein-labelled wt DNA with ORF56.
Figure 7C illustrates a titration under stoichiometric binding conditions (wt DNA concentration 50 nM). The intersection of the two linear parts of the binding isotherm yields stoichiometric equivalence at 220 nM protein monomer concentration, suggesting that dimeric ORF56 forms a tetramer on the DNA.
Our titration curves (e.g. Fig. 7A) did not indicate any cooperativity in binding of the two dimers. We therefore considered two binding models for evaluation of the titration curves. In one model binding of two dimers to two identical binding sites, the two half-sites, was assumed. The other model is based on binding of a preformed tetramer of ORF56 to the inverted repeat. Binding constants evaluated from the two models will differ by a factor of two.
A DNA probe containing one half-site of the inverted repeat was bound with low affinity (see Table 2) indicating that a model with two identical binding sites is inappropriate. We therefore fitted our data according to the tetramer binding model and the binding constants given in Table 2 have been calculated on the basis of an ORF56 concentration that is one quarter of the monomer concentration. The tetramer binding model, however, contradicts our gel filtration data, which do not indicate the presence of free tetramers of ORF56. Therefore, the exact mechanism of ORF56 DNA binding remains to be established (see Discussion).
Table 2. Dissociation constants of ORF56 as measured by fluorescence anisotropy titrations.
| DNA |
Buffer |
Kd (nM) |
Kc (nM)a |
Specificityb |
| wt-F | Tris–HCl | 1.5 (0.63–2.45)c | ||
| Na cacodylate | 0.4d | |||
| wt | Tris–HCl | 0.88 ± 0.11e | ||
| rep | Tris–HCl | 210 ± 13f | ||
| CT DNA | Tris–HCl | 670 ± 70 | 270 | |
| CT DNA (53°C) | Tris–HCl | 1220 ± 100 | 1500 |
The buffer in all experiments was 20 mM Tris–HCl, pH 7.5, 100 mM KCl, 0.01% Tween 20. 20 mM Tris–HCl was substituted for 20 mM sodium cacodylate where indicated. wt-F is the fluorescently labelled wt DNA. Rep is a single repeat DNA, CT DNA is sonicated calf thymus DNA. Kd is the dissociation constant of the equilibrium (ORF56)4 + DNA ⇔ DNA·(ORF56)4. The Kd value was obtained by least square fitting of the binding isotherm and refers to the tetramer concentration.
aDetermined from a competition experiment. The dissociation constant Kc refers to the microscopic dissociation constant for all non-specific sites on calf thymus DNA.
bSpecificity was calculated by Kc/Kd. Kd at 25°C was 2.45 ± 0.23 nM and at 53°C 0.8 ± 0.2 nM.
cRange of six independent experiments.
dAverage of two experiments.
eDetermined from a competition assay.
fDetermined from a competition assay. The dissociation constant applies to a dimer half-site equilibrium.
Competition experiments to determine the specificity of binding
In order to quantify the specificity of binding of ORF56 to DNA we performed competition experiments with calf thymus DNA. For these experiments we preformed a complex of protein with fluorescently labelled wt DNA. Increasing amounts of competitor DNA were then added and the decrease in anisotropy was recorded. A large excess of non-specific DNA was needed to displace ORF56 from the cognate DNA (Fig. 8A). At 25°C we found that non-specific DNA is bound with a microscopic dissociation constant of 670 nM, which is ∼270-fold weaker than specific binding. At 53°C, which is closer to the physiological temperature of S.islandicus, the specificity ratio is ∼1500 (Table 2).
Figure 8.



Specificity, salt and temperature dependance of the ORF56 DNA-binding activity. (A) Competition experiments with non-specific DNA. ORF56 (20 nM) was allowed to bind to 2 nM fluorescently labelled wt DNA. Afterwards increasing amounts of competitor DNA were added. Filled squares, competition with rep DNA (single repeat substrate); open circles, competition with calf thymus DNA at 25°C; filled circles, competition with calf thymus DNA at 53°C. The calf thymus DNA concentration refers to the concentration of base pairs, which equals the concentration of overlapping sites. (B) Salt dependence of the dissociation constant. wt DNA (2 nM) was titrated in 10 mM Tris–HCl, pH 7.5, 0.01% Tween 20 with 125–300 mM KCl. The dissociation constant Kd at each salt concentration was calculated and plotted as a function of added salt. The calculated slope of the plot is 6.8 ± 0.2. (C) Temperature dependence of DNA binding. The temperature dependence of the dissociation constant was assayed at eight temperatures in the range 17–57°C. Fluorescently labelled wt DNA (5 nM) was titrated in cacodylate buffer with 150 mM KCl. The dissociation constants Kd were calculated at each temperature and plotted as lnKd versus 1/T (van’t Hoff plot). The data was fitted according to equation 4 (see Materials and Methods). ΔCp = –6.2 kJ/mol, TH = 39°C and TS = 47°C.
We also used the competition assay to assess ORF56 binding to the half-site of its target DNA. For these experiments we used a DNA probe comprising a single repeat from the cognate wt DNA as competitor. A large excess of rep DNA was necessary to reduce the amount of ORF56 wt DNA complex, indicating that binding of ORF56 to the single repeat is rather weak. Quantitative analysis of the isotherms yields a dissociation constant of 210 nM for the ORF56 dimer and rep DNA equilibria. With the tetramer binding model the dissociation constant is 105 nM, which is large compared to the dissociation constant of the ORF56 tetramer and wt DNA equilibrium, which was 2.2 nM in this particular experiment. The low affinity for the half-site clearly supports the tetramer binding model.
Salt and temperature dependence of DNA binding
Figure 8B illustrates the salt dependence of the dissociation constant. The salt dependence of protein–DNA interactions is generally explained by ion release upon binding (21,22). The released ions make an entropic contribution which decreases at higher salt concentrations. The slope of the double logarithmic plot is related to the number of ions released according to the general formula:
No. of ions released = (∂logKobs)/(∂log[salt])
For binding of the ORF56 tetramer to the inverted repeat the slope is 6.8, indicating that about seven ions are released upon complexation.
The temperature dependence of binding was analysed by anisotropy titrations in the temperature range 17–57°C. van’t Hoff analysis of the data reveals a non-linear relationship (Fig. 8C). Assuming that the change in heat capacity (ΔCp) is invariant over the temperature range investigated the data can be fitted to equation 4. We obtain TH = 39°C, TS = 47°C and ΔCp = –6.2 kJ/mol. TH is the temperature at which ΔH = 0 and Kd is at a minimum. TS is the temperature at which ΔS = 0 and the free energy change, ΔG, is minimal. The highest temperature investigated was 57°C, however, this measurement could not be included in the global fit with the other binding isotherms (see Materials and Methods). Therefore, the relative error at this temperature is quite high. As the wt DNA probe melts at 70°C in standard buffer we could not extend our measurements to higher temperatures. The optimal growth temperature of S.islandicus is ∼80°C (23). This contrasts with the strongest binding observed at 39°C. However, DNA melting experiments in the presence of ORF56 clearly show that a stable DNA–protein complex is also formed at much higher temperatures.
ORF56 protects double-stranded DNA from heat denaturation
ORF56 is highly thermostable. Up to 100°C there is no denaturation of the α-helical structure, as can be seen in the CD temperature profile at 222 nm (data not shown). A 10 min incubation at 100°C weakened its DNA-binding activity by only ∼35%. The high thermostability of ORF56 enabled us to investigate whether ORF56 protects DNA from heat denaturation. The melting curve of wt+GCG DNA was monitored at 260 nm in the absence and presence of stoichiometric amounts of ORF56 (Fig. 9). The DNA was stabilised by ∼15°C upon complexation with ORF56, indicating that melting of the protein-bound double-stranded DNA requires more energy. This energy is needed to dissolve the double-stranded DNA–protein complex as ORF56 does not bind to single-stranded DNA. The DNA denaturation temperature profiles also demonstrate that, even at temperatures as high as 85°C, ORF56 is able to bind DNA.
Figure 9.

Thermal denaturation of wt+CGC DNA in the absence and presence of ORF56. The temperature profiles of 1 µM wt+CGC DNA (open circles) and of 1 µM wt+CGC DNA and 4 µM ORF56 (closed circles) are shown. Every second data point is presented. The melting temperature Tm was obtained by fitting according to equation 5 (see Materials and Methods). Tm was 71 and 86°C, respectively.
DISCUSSION
In the present study we report on the DNA-binding properties of the ORF56 protein encoded by plasmid pRN1 from S.islandicus. By probing recombinant ORF56 with different DNA substrates we have shown that ORF56 binds specifically to an inverted repeat sequence located upstream of its own initiator codon.
Analysis of Sulfolobus ssp. promoter sequences revealed three consensus sequences upstream of the initiator methionine codon (24,25): a BRE (transcription factor B-responsive element) with consensus (A/G)N(A/T)AA(A/T) immediately upstream of an archael TATA box with consensus sequence (T/C)TT(A/T)(A/T)A(A/T)(A/T) and a box B proximal to the initiation site with consensus sequence (T/A)TG(A/C). All three boxes are found in the upstream region of the orf56 gene (Fig. 5). Box B is located within the inverted repeat. By binding to this region ORF56 could interfere with assembly of the archael transcription apparatus and thereby repress its own transcription.
The gene orf56 overlaps with gene orf904, which codes for the putative initiator protein of plasmid replication. Sequence analysis does not reveal a typical promoter sequence upstream of the orf904 gene. Possibly, orf56 and orf904 are co-transcribed from the promoter upstream of orf56. A model where ORF56 negatively regulates both its own synthesis and synthesis of the putative initiator protein seems plausible. This type of feedback regulation of initiator protein synthesis has been described for the eubacterial rolling circle plasmids pLS1 and pE194 (26,27), where the CopG proteins function as negative regulators.
ORF56 shows sequence similiarity to the CopG proteins (see Fig. 1), the DNA-binding sites, however, are not related. The putative initiator protein ORF904 from pRN1 does not show sequence similiarity to the initiator proteins from the eubacterial plasmids pLS1 and pE194, which are 30% identical at the amino acid level. It is therefore possible that the copy control mechanisms of the eubacterial and archael plasmids have evolved independently.
The experimental data for binding to wt DNA is well fitted by a binding model where two dimers of ORF56 are bound to one inverted repeat on the DNA. This model is in line with the crystal structure of the homologous CopG protein from pLS1 in complex with DNA, where two CopG dimers are bound to an inverted repeat (19).
We did not observe any cooperativity in the binding isotherms. Furthermore, DNA containing a half-site of the inverted repeat is very weakly bound in EMSA and competitive fluorescence anisotropy measurements. We therefore propose a binding scheme where a preformed ORF56 tetramer reacts with the DNA.
A different oligomerisation behaviour has been reported for two well-studied ribbon helix–turn–helix proteins, namely the bacteriophage P22 arc and mnt repressors. The arc repressor is monomeric and unfolded at low protein concentrations and folds and dimerises in a two-state reaction to DNA-binding competent dimers. One dimer then binds to an operator half-site and the second dimer binds with increased affinity to the second half of the operator. The cooperativity of the binding reaction is therefore very high and was determined to be ∼3.5 (28). In contrast, the homologous and structurally related mnt repressor is tetrameric in solution and binds its operator obeying a simple one-site model with no cooperativity (29).
Our data indicate that ORF56 is dimeric in solution but forms a tetramer on the DNA with no obvious cooperativity in the binding isotherms. A similar DNA-promoted oligomerisation has been reported for the monomeric bacteriophage 434 repressor, which dimerises in the presence of specific and non-specific DNA and is competent for sequence-specific binding in its dimeric state (30). The dimer–tetramer equilibrium of ORF56 might show a comparable behaviour. Clearly, more data, i.e. kinetic experiments, are necessary to elucidate the assembly and DNA recognition pathway of ORF56.
Our study of the temperature dependence of DNA binding shows that the highest binding affinity is achieved at ∼40°C. Direct determination of the dissociation constant at 80°C, the optimal growth temperature of S.islandicus, was not possible as the wt DNA substrate melts at 70°C. van’t Hoff analysis revealed a large negative change in heat capacity (ΔCp = –6.2 kJ/mol) which is comparable to the heat capacity change observed for other DNA-binding proteins, as for example the lac repressor and the EcoRI endonuclease, for which ΔCp is in the range –4 to –5 kJ/mol. For these proteins the negative heat capacity change on association was largely attributed to burial of a non-polar surface area upon specific binding and concomitant folding (11,22,31). Burial of non-polar surfaces is thought to be a general feature of sequence-specific DNA-binding proteins. Specificity is ensured in this process by the complementarity of the surfaces of the specifically bound DNA and the protein. Complex formation releases surface bound water to the bulk water, making an entropically favourable energetic contribution.
Furthermore, our CD data show that ORF56 becomes more structured upon DNA binding. This could also make a contribution to the observed negative heat capacity change upon association.
The data presented here suggest that ORF56 adopts the same fold as the CopG protein, which is a ribbon helix–turn–helix protein. Both proteins are dimeric in solution and form a tetramer on the DNA, where they recognise an inverted repeat. Furthermore, preliminary 2- and 3-dimensional NMR data from our laboratory suggest that residues Gly2–Leu8 and Asp53–Lys56 are unstructured in solution in the absence of DNA. We speculate that the termini become structured upon binding, leading to the change in CD signal. The decrease in ellipticity in the region 200–230 nm can be interpreted as additional formation of α-helices or type II β-turns. As both termini are positively charged they could form salt bridges with the phosphates of the DNA upon binding and become concomitantly structured. It is noteworthy that in the case of the arc repressor, which is a ribbon helix–turn–helix protein, formation of β-turns is induced at the N-terminus upon binding (32).
Our study reveals that optimal binding of operator DNA occurs at ∼40°C, which is far below the optimal growth temperature of S.islandicus. So far no sequence-specific thermophilic DNA-binding protein has been studied thermodynamically. Only the DNA-binding properties of the histone-like Sso7d protein from S.solfataricus has been investigated thoroughly. The temperature dependence of its non-specific binding to poly(dG·dC) has been studied in the temperature range 15–45°C (33). Extrapolation of the data to higher temperatures indicated that optimal binding could well be at ∼80–100°C.
Extrapolation of our data to higher temperatures yields a dissociation constant Kd of ∼1 µM at 80°C. However, this value is considered to be very speculative given the large range of extrapolation. Indeed, the DNA heat denaturing experiment clearly shows that even at 85°C there is substantial binding of ORF56 to its cognate DNA. Further studies will be necessary to clarify how ORF56 recognises its target DNA at ∼80°C, its physiological temperature.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. W.F.Doolittle (Dalhousie University, Halifax) for plasmid pUC18-pRN1. We also appreciate the support of our colleagues J.Balbach and T.Hey for critically reading the manuscript.
References
- 1.Schleper C., Kubo,K. and Zillig,W. (1992) The particle SSV1 from the extremely thermophilic archaeon Sulfolobus is a virus: demonstration of infectivity and of transfection with viral DNA. Proc. Natl Acad. Sci. USA, 89, 7645–7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stedman K.M., Schleper,C., Rumpf,E. and Zillig,W. (1999) Genetic requirements for the function of the archaeal virus SSV1 in Sulfolobus solfataricus: construction and testing of viral shuttle vectors. Genetics, 152, 1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zillig W., Kletzin,A., Schleper,C., Holz,I., Janekovic,D., Hain,J., Lanzendoerfer,M. and Kristjansson,J.K. (1994) Screening for Sulfolobales, their plasmids and their viruses in Icelandic solfataras. Syst. Appl. Microbiol., 16, 609–628. [Google Scholar]
- 4.Keeling P.J., Klenk,H.P., Singh,R.K., Feeley,O., Schleper,C., Zillig,W., Doolittle,W.F. and Sensen,C.W. (1996) Complete nucleotide sequence of the Sulfolobus islandicus multicopy plasmid pRN1. Plasmid, 35, 141–144. [DOI] [PubMed] [Google Scholar]
- 5.Peng X., Holz,I., Zillig,W., Garrett,R.A. and She,Q. (2000) Evolution of the family of pRN plasmids and their integrase-mediated insertion into the chromosome of the crenarchaeon Sulfolobus solfataricus. J. Mol. Biol., 303, 449–454. [DOI] [PubMed] [Google Scholar]
- 6.Keeling P.J., Klenk,H.P., Singh,R.K., Schenk,M.E., Sensen,C.W., Zillig,W. and Doolittle,W.F. (1998) Sulfolobus islandicus plasmids pRN1 and pRN2 share distant but common evolutionary ancestry. Extremophiles, 2, 391–393. [DOI] [PubMed] [Google Scholar]
- 7.Arnold H.P., She,Q., Phan,H., Stedman,K., Prangishvili,D., Holz,I., Kristjansson,J.K., Garrett,R. and Zillig,W. (1999) The genetic element pSSVx of the extremely thermophilic crenarchaeon sulfolobus is a hybrid between a plasmid and a virus. Mol. Microbiol., 34, 217–226. [DOI] [PubMed] [Google Scholar]
- 8.Kletzin A., Lieke,A., Urich,T., Charlebois,R.L. and Sensen,C.W. (1999) Molecular analysis of pDL10 from Acidianus ambivalens reveals a family of related plasmids from extremely thermophilic and acidophilic archaea. Genetics, 152, 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espinosa M., del Solar,G.H., Rojo,F. and Alonso,J.C. (1995) Plasmid rolling circle replication and its control. FEMS Microbiol. Lett., 130, 111–120. [DOI] [PubMed] [Google Scholar]
- 10.Khan S.A. (1997) Rolling-circle replication of bacterial plasmids. Microbiol. Mol. Biol. Rev., 61, 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha J.H., Spolar,R.S. and Record,M.T.J. (1989) Role of the hydrophobic effect in stability of site-specific protein–DNA complexes. J. Mol. Biol., 209, 801–816. [DOI] [PubMed] [Google Scholar]
- 12.Schagger H. and von Jagow,G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem., 166, 368–379. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin R.L. (1986) Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl Acad. Sci. USA, 83, 8069–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem., 237, 260–273. [DOI] [PubMed] [Google Scholar]
- 15.Puglisi J.D. and Tinoco,I.J. (1989) Absorbance melting curves of RNA. Methods Enzymol., 180, 304–325. [DOI] [PubMed] [Google Scholar]
- 16.Bohm G., Muhr,R. and Jaenicke,R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng., 5, 191–195. [DOI] [PubMed] [Google Scholar]
- 17.Garnier J., Gibrat,J.F. and Robson,B. (1996) GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol., 266, 540–553. [DOI] [PubMed] [Google Scholar]
- 18.Woody R.W. (1995) Circular dichroism. Methods Enzymol., 246, 34–71. [DOI] [PubMed] [Google Scholar]
- 19.Gomis R.F., Sola,M., Acebo,P., Parraga,A., Guasch,A., Eritja,R., Gonzalez,A., Espinosa,M., del-Solar,G. and Coll,M. (1998) The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J., 17, 7404–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eftink M.R. (1997) Fluorescence methods for studying equilibrium macromolecule–ligand interactions. Methods Enzymol., 278, 221–257. [DOI] [PubMed] [Google Scholar]
- 21.Record M.T.J., Ha,J.H. and Fisher,M.A. (1991) Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol., 208, 291–343. [DOI] [PubMed] [Google Scholar]
- 22.Hard T. and Lundback,T. (1996) Thermodynamics of sequence-specific protein–DNA interactions. Biophys. Chem., 62, 121–139. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Garcia P. and Forterre,P. (1999) Control of DNA topology during thermal stress in hyperthermophilic archaea: DNA topoisomerase levels, activities and induced thermotolerance during heat and cold shock in Sulfolobus. Mol. Microbiol., 33, 766–777. [DOI] [PubMed] [Google Scholar]
- 24.Reiter W.D., Palm,P. and Zillig,W. (1988) Analysis of transcription in the archaebacterium Sulfolobus indicates that archaebacterial promoters are homologous to eukaryotic pol II promoters. Nucleic Acids Res., 16, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell S.D., Kosa,P.L., Sigler,P.B. and Jackson,S.P. (1999) Orientation of the transcription preinitiation complex in archaea. Proc. Natl Acad. Sci. USA, 96, 13662–13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.del Solar G.H., Perez,M.J. and Espinosa,M. (1990) Plasmid pLS1-encoded RepA protein regulates transcription from repAB promoter by binding to a DNA sequence containing a 13-base pair symmetric element. J. Biol. Chem., 265, 12569–12575. [PubMed] [Google Scholar]
- 27.Kwak J.H. and Weisblum,B. (1994) Regulation of plasmid pE194 replication: control of cop-repF operon transcription by Cop and of repF translation by countertranscript RNA. J. Bacteriol ., 176, 5044–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown B.M., Bowie,J.U. and Sauer,R.T. (1990) Arc repressor is tetrameric when bound to operator DNA. Biochemistry, 29, 11189–11195. [DOI] [PubMed] [Google Scholar]
- 29.Knight K.L., Bowie,J.U., Vershon,A.K., Kelley,R.D. and Sauer,R.T. (1989) The Arc and Mnt repressors. A new class of sequence-specific DNA-binding protein. J. Biol. Chem., 264, 3639–3642. [PubMed] [Google Scholar]
- 30.Ciubotaru M., Bright,F.V., Ingersoll,C.M. and Koudelka,G.B. (1999) DNA-induced conformational changes in bacteriophage 434 repressor. J. Mol. Biol., 294, 859–873. [DOI] [PubMed] [Google Scholar]
- 31.Spolar R.S. and Record,M.T.J. (1994) Coupling of local folding to site-specific binding of proteins to DNA. Science, 263, 777–784. [DOI] [PubMed] [Google Scholar]
- 32.Raumann B.E., Rould,M.A., Pabo,C.O. and Sauer,R.T. (1994) DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature, 367, 754–757. [DOI] [PubMed] [Google Scholar]
- 33.Lundback T., Hansson,H., Knapp,S., Ladenstein,R. and Hard,T. (1998) Thermodynamic characterization of non-sequence-specific DNA-binding by the Sso7d protein from Sulfolobus solfataricus. J. Mol. Biol., 276, 775–786. [DOI] [PubMed] [Google Scholar]
- 34.Qureshi S.A. and Jackson,S.P. (1998) Sequence-specific DNA binding by the S. shibatae TFIIB homolog, TFB and its effect on promoter strength. Mol. Cell, 1, 389–400. [DOI] [PubMed] [Google Scholar]