

Abstract
The crystal structure of the title compound, C15H13N3O, consists of columns of molecules that are interconnected by N—H⋯N hydrogen bonds in the direction of the b axis. The torsion angle between the imidazo[1,2-a]pyridine ring system and the phenyl ring is 9.04 (5)°.
Related literature
For general background, see Anaflous et al. (2004 ▶); Gueffier et al. (1998 ▶); Mavel et al. (2002 ▶).
Experimental
Crystal data
C15H13N3O
M r = 251.3
Monoclinic,
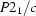
a = 13.9680 (5) Å
b = 5.6784 (2) Å
c = 15.8145 (5) Å
β = 101.039 (3)°
V = 1231.13 (7) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 120 K
0.58 × 0.25 × 0.17 mm
Data collection
Oxford Diffraction Xcalibur2 diffractometer with Sapphire2 CCD detector
Absorption correction: none
15703 measured reflections
2556 independent reflections
1544 reflections with I > 3σ(I)
R int = 0.054
Refinement
R[F 2 > 2σ(F 2)] = 0.036
wR(F 2) = 0.084
S = 1.00
2556 reflections
175 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.17 e Å−3
Δρmin = −0.14 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2008 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2008 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SIR2002 (Burla et al., 2003 ▶); program(s) used to refine structure: JANA2006 (Petříček et al., 2006 ▶); molecular graphics: DIAMOND (Brandenburg & Putz, 1999 ▶); software used to prepare material for publication: JANA2006.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808011501/fj2112sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011501/fj2112Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N3—H3n⋯N1i | 0.880 (12) | 2.162 (12) | 3.0219 (16) | 165.4 (13) |
Symmetry code: (i) 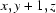 .
.
Acknowledgments
We thank the Grant Agency of the Czech Republic for support, grant No. 202/05/0757.
supplementary crystallographic information
Comment
In recent years, functionalized imidazo[1,2-a]pyridine and imidazo[1,2-a]pyrimidine systems attracted persistent interest due to their biological activities (Anaflous et al., 2004 and reference herein). The screening of imidazo[1,2-a]pyridine derivatives against tuberculosis showed interesting results (Anaflous et al., 2004) and many functionalized imidazo[1,2-a]pyridines bearing a thioether side chain at the 3 position are reported as highly active against human cytomegalovirus and /or varicella-zoster virus (Gueffier et al., 1998 & Mavel et al., 2002).
We report in the present paper on the synthesis and crystal structure of N-(2-phenylimidazo[1,2-a]pyridin-3-yl)acetamide (I).
The molecules of the title compound are interconnected into columns extended along b by an N3—H3n···N1 hydrogen bonds (see Tab. 1). No bonding has been found between the columns that appear to be quite isolated.
Bonds and angles values are usual as those reported in similar compounds.
The torsion angle between the imidazo[1,2-a]pyridine and phenyl ring is 9.04 (5)°
Experimental
The commercially available 2-phenylimidazo[1,2-a]pyridin-3-amine (0.50 g, 2.4 mmole) in toluene (10 ml, 94 mmole) was treated with acetic anhydride (0.3 ml, 3.2 mmole). The mixture was stirred for two hours. Toluene was eliminated under reduced pressure and the residue was washed with water to give, after drying, 0.45 g (1.8 mmole) of N-(2-phenylimidazo[1,2-a]pyridin-3-yl)acetamide as colorless crystals.
Figures
Fig. 1.

View of the unit cell of the title structure along the axis b
Fig. 2.

Asymetric unit of title compound, showing 50% displacement ellispoids for non-H atoms.
Fig. 3.

The columns of molecules showing N—H···N hydrogen bonds
Crystal data
| C15H13N3O | F000 = 528 |
| Mr = 251.3 | Dx = 1.355 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 4755 reflections |
| a = 13.9680 (5) Å | θ = 2.6–26.5º |
| b = 5.6784 (2) Å | µ = 0.09 mm−1 |
| c = 15.8145 (5) Å | T = 120 K |
| β = 101.039 (3)º | Prism, colorless |
| V = 1231.13 (7) Å3 | 0.58 × 0.25 × 0.17 mm |
| Z = 4 |
Data collection
| Oxford Diffraction Xcalibur2 diffractometer with Sapphire2 CCD detector | 2556 independent reflections |
| Radiation source: X-ray tube | 1544 reflections with I > 3σ(I) |
| Monochromator: graphite | Rint = 0.054 |
| Detector resolution: 8.3438 pixels mm-1 | θmax = 26.5º |
| T = 120 K | θmin = 2.6º |
| Rotation method data acquisition using ω scans | h = −17→17 |
| Absorption correction: none | k = −7→7 |
| 15703 measured reflections | l = −19→19 |
Refinement
| Refinement on F2 | 45 constraints |
| R[F2 > 2σ(F2)] = 0.036 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.084 | Weighting scheme based on measured s.u.'s w = 1/[σ2(I) + 0.0016I2] |
| S = 1.00 | (Δ/σ)max = 0.006 |
| 2556 reflections | Δρmax = 0.17 e Å−3 |
| 175 parameters | Δρmin = −0.14 e Å−3 |
| 1 restraint | Extinction correction: none |
Special details
| Refinement. The refinement was carried out against all reflections. The conventional R-factor is always based on F. The goodness of fit as well as the weighted R-factor are based on F and F2 for refinement carried out on F and F2, respectively. The threshold expression is used only for calculating R-factors etc. and it is not relevant to the choice of reflections for refinement.All the H atoms were discernible in difference Fourier maps and could be refined to reasonable geometry. According to standard procedures for organic compounds the H atoms bonded to C atoms were constrained to ideal positions. The N—H distances were restrained to 0.87 Å with σ 0.01. The isotropic atomic displacement parameters of hydrogen atoms were evaluated as 1.2*Ueq of the parent atom.The program used for refinement, Jana2006, uses the weighting scheme based on the experimental expectations, see _refine_ls_weighting_details, that does not force S to be one. Therefore the values of S are usually larger than the ones from the SHELX program. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.61764 (8) | −0.2561 (2) | 0.47985 (7) | 0.0230 (4) | |
| N2 | 0.56626 (7) | 0.05089 (18) | 0.39364 (7) | 0.0206 (4) | |
| N3 | 0.70718 (11) | 0.3084 (2) | 0.41732 (9) | 0.0235 (5) | |
| O1 | 0.79160 (9) | 0.1417 (2) | 0.32433 (8) | 0.0298 (4) | |
| C1 | 0.69090 (11) | −0.0912 (2) | 0.48648 (9) | 0.0210 (5) | |
| C2 | 0.66186 (9) | 0.09842 (19) | 0.43398 (8) | 0.0224 (4) | |
| C3 | 0.54173 (10) | −0.1658 (2) | 0.42431 (9) | 0.0211 (5) | |
| C4 | 0.44673 (11) | −0.2519 (3) | 0.39578 (9) | 0.0238 (5) | |
| C5 | 0.38234 (11) | −0.1217 (3) | 0.33930 (9) | 0.0262 (5) | |
| C6 | 0.41070 (11) | 0.0979 (2) | 0.30893 (9) | 0.0272 (5) | |
| C7 | 0.50162 (10) | 0.1807 (3) | 0.33604 (9) | 0.0233 (5) | |
| C8 | 0.78417 (11) | −0.1346 (2) | 0.54688 (10) | 0.0220 (6) | |
| C9 | 0.79074 (12) | −0.3221 (3) | 0.60385 (10) | 0.0285 (5) | |
| C10 | 0.87592 (12) | −0.3699 (3) | 0.66086 (10) | 0.0317 (5) | |
| C11 | 0.95689 (11) | −0.2316 (3) | 0.66266 (10) | 0.0290 (5) | |
| C12 | 0.95210 (12) | −0.0434 (3) | 0.60733 (10) | 0.0373 (6) | |
| C13 | 0.86634 (12) | 0.0062 (3) | 0.54974 (11) | 0.0356 (6) | |
| C14 | 0.77211 (11) | 0.3171 (3) | 0.36247 (10) | 0.0220 (5) | |
| C15 | 0.81873 (12) | 0.5518 (3) | 0.35553 (11) | 0.0294 (6) | |
| H3n | 0.6911 (10) | 0.4370 (19) | 0.4423 (9) | 0.0282* | |
| H4 | 0.42756 | −0.400812 | 0.415958 | 0.0285* | |
| H5 | 0.317152 | −0.17869 | 0.319756 | 0.0314* | |
| H6 | 0.364663 | 0.187888 | 0.268776 | 0.0327* | |
| H7 | 0.520896 | 0.328874 | 0.315284 | 0.028* | |
| H9 | 0.734759 | −0.420445 | 0.603502 | 0.0341* | |
| H10 | 0.878784 | −0.500882 | 0.699648 | 0.038* | |
| H11 | 1.016393 | −0.265961 | 0.702246 | 0.0348* | |
| H12 | 1.008436 | 0.054304 | 0.608524 | 0.0447* | |
| H13 | 0.863646 | 0.13862 | 0.511608 | 0.0427* | |
| H15a | 0.77381 | 0.67451 | 0.36305 | 0.0353* | |
| H15b | 0.876474 | 0.564915 | 0.399312 | 0.0353* | |
| H15c | 0.8358 (10) | 0.5666 (19) | 0.2998 (9) | 0.0353* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0273 (7) | 0.0188 (6) | 0.0250 (7) | −0.0018 (5) | 0.0100 (6) | −0.0017 (5) |
| N2 | 0.0230 (7) | 0.0198 (6) | 0.0205 (6) | 0.0005 (5) | 0.0079 (5) | −0.0023 (5) |
| N3 | 0.0343 (9) | 0.0126 (7) | 0.0271 (8) | −0.0014 (7) | 0.0148 (7) | −0.0020 (7) |
| O1 | 0.0405 (7) | 0.0228 (6) | 0.0305 (7) | 0.0012 (6) | 0.0176 (6) | −0.0026 (5) |
| C1 | 0.0263 (9) | 0.0171 (7) | 0.0225 (8) | −0.0029 (7) | 0.0119 (7) | −0.0042 (6) |
| C2 | 0.0267 (8) | 0.0183 (6) | 0.0245 (7) | −0.0015 (6) | 0.0108 (6) | −0.0028 (5) |
| C3 | 0.0275 (9) | 0.0186 (7) | 0.0192 (8) | 0.0014 (7) | 0.0098 (7) | −0.0021 (6) |
| C4 | 0.0304 (9) | 0.0210 (8) | 0.0227 (8) | −0.0023 (7) | 0.0120 (7) | −0.0040 (6) |
| C5 | 0.0217 (9) | 0.0333 (8) | 0.0247 (8) | −0.0023 (7) | 0.0074 (7) | −0.0094 (7) |
| C6 | 0.0317 (8) | 0.0309 (8) | 0.0195 (8) | 0.0080 (6) | 0.0062 (7) | 0.0003 (6) |
| C7 | 0.0319 (8) | 0.0197 (8) | 0.0207 (8) | 0.0027 (7) | 0.0112 (7) | −0.0005 (6) |
| C8 | 0.0226 (10) | 0.0222 (8) | 0.0222 (9) | 0.0012 (7) | 0.0068 (8) | −0.0058 (7) |
| C9 | 0.0296 (9) | 0.0261 (9) | 0.0308 (9) | −0.0033 (8) | 0.0086 (7) | 0.0015 (7) |
| C10 | 0.0338 (9) | 0.0285 (8) | 0.0327 (9) | 0.0036 (7) | 0.0061 (8) | 0.0048 (7) |
| C11 | 0.0282 (8) | 0.0324 (9) | 0.0258 (8) | 0.0039 (7) | 0.0034 (7) | −0.0015 (7) |
| C12 | 0.0293 (10) | 0.0377 (9) | 0.0425 (10) | −0.0114 (8) | 0.0013 (8) | 0.0022 (7) |
| C13 | 0.0394 (10) | 0.0306 (9) | 0.0357 (10) | −0.0038 (8) | 0.0040 (8) | 0.0111 (8) |
| C14 | 0.0251 (10) | 0.0210 (9) | 0.0203 (8) | 0.0030 (7) | 0.0055 (7) | 0.0034 (7) |
| C15 | 0.0335 (10) | 0.0248 (10) | 0.0331 (11) | −0.0032 (8) | 0.0144 (9) | 0.0020 (9) |
Geometric parameters (Å, °)
| N1—C1 | 1.3763 (18) | C6—H6 | 0.96 |
| N1—C3 | 1.3424 (17) | C7—H7 | 0.96 |
| N2—C2 | 1.3916 (15) | C8—C9 | 1.387 (2) |
| N2—C3 | 1.3894 (17) | C8—C13 | 1.393 (2) |
| N2—C7 | 1.3689 (16) | C9—C10 | 1.375 (2) |
| N3—C2 | 1.3984 (19) | C9—H9 | 0.96 |
| N3—C14 | 1.371 (2) | C10—C11 | 1.373 (2) |
| N3—H3n | 0.880 (12) | C10—H10 | 0.96 |
| O1—C14 | 1.222 (2) | C11—C12 | 1.375 (2) |
| C1—C2 | 1.3727 (18) | C11—H11 | 0.96 |
| C1—C8 | 1.481 (2) | C12—C13 | 1.388 (2) |
| C3—C4 | 1.405 (2) | C12—H12 | 0.96 |
| C4—C5 | 1.359 (2) | C13—H13 | 0.96 |
| C4—H4 | 0.96 | C14—C15 | 1.497 (2) |
| C5—C6 | 1.419 (2) | C15—H15a | 0.96 |
| C5—H5 | 0.96 | C15—H15b | 0.96 |
| C6—C7 | 1.345 (2) | C15—H15c | 0.960 (15) |
| C1—N1—C3 | 105.82 (11) | C1—C8—C9 | 119.17 (14) |
| C2—N2—C3 | 106.91 (10) | C1—C8—C13 | 122.90 (14) |
| C2—N2—C7 | 130.80 (11) | C9—C8—C13 | 117.93 (14) |
| C3—N2—C7 | 122.27 (11) | C8—C9—C10 | 121.26 (15) |
| C2—N3—C14 | 121.89 (13) | C8—C9—H9 | 119.372 |
| C2—N3—H3n | 117.3 (9) | C10—C9—H9 | 119.371 |
| C14—N3—H3n | 120.8 (9) | C9—C10—C11 | 120.43 (14) |
| N1—C1—C2 | 110.99 (11) | C9—C10—H10 | 119.784 |
| N1—C1—C8 | 119.03 (12) | C11—C10—H10 | 119.784 |
| C2—C1—C8 | 129.96 (13) | C10—C11—C12 | 119.48 (13) |
| N2—C2—N3 | 120.55 (11) | C10—C11—H11 | 120.26 |
| N2—C2—C1 | 105.77 (11) | C12—C11—H11 | 120.259 |
| N3—C2—C1 | 133.68 (12) | C11—C12—C13 | 120.42 (15) |
| N1—C3—N2 | 110.49 (11) | C11—C12—H12 | 119.792 |
| N1—C3—C4 | 131.02 (13) | C13—C12—H12 | 119.792 |
| N2—C3—C4 | 118.48 (12) | C8—C13—C12 | 120.48 (15) |
| C3—C4—C5 | 119.18 (14) | C8—C13—H13 | 119.758 |
| C3—C4—H4 | 120.41 | C12—C13—H13 | 119.758 |
| C5—C4—H4 | 120.41 | N3—C14—O1 | 121.32 (15) |
| C4—C5—C6 | 120.52 (13) | N3—C14—C15 | 115.37 (14) |
| C4—C5—H5 | 119.739 | O1—C14—C15 | 123.28 (16) |
| C6—C5—H5 | 119.74 | C14—C15—H15a | 109.471 |
| C5—C6—C7 | 120.46 (13) | C14—C15—H15b | 109.472 |
| C5—C6—H6 | 119.768 | C14—C15—H15c | 109.5 (7) |
| C7—C6—H6 | 119.769 | H15a—C15—H15b | 109.472 |
| N2—C7—C6 | 119.07 (13) | H15a—C15—H15c | 109.471 |
| N2—C7—H7 | 120.466 | H15b—C15—H15c | 109.47 |
| C6—C7—H7 | 120.465 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3n···N1i | 0.880 (12) | 2.162 (12) | 3.0219 (16) | 165.4 (13) |
Symmetry codes: (i) x, y+1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2112).
References
- Anaflous, A., Benchat, N., Mimouni, M., Abouricha, S., Ben-Hadda, T., El Bali, B., Hakkou, A. & Hacht, B. (2004). Lett. Drug Des. Discovery, 1, 224—229.
- Brandenburg, K. & Putz, H. (1999). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Burla, M. C., Camalli, M., Carrozzini, B., Cascarano, G. L., Giacovazzo, C., Polidori, G. & Spagna, R. (2003). J. Appl. Cryst.36, 1103.
- Gueffier, A., Mavel, S., Lhassani, M., Elhakmaoui, A., Snoeck, R., Andrei, G., Chavignon, O., Teulade, J. C., Witvrouw, M., Balzarini, J., De Clercq, E. & Chapat, J. (1998). J. Med. Chem.41, 5108–5112. [DOI] [PubMed]
- Mavel, S., Renou, J. L., Galtier, C., Allouchi, H., Snoeck, R., Andrei, G., Balzarini, J., Gueffier, A. & De Clercq, E. (2002). Bioorg. Med Chem.10, 941–946. [DOI] [PubMed]
- Oxford Diffraction (2008). CrysAlis CCD andCrysAlis RED Oxford Diffraction Ltd., Abingdon, Oxfordshire, England.
- Petříček, V., Dušek, M. & Palatinus, L. (2006). JANA2006 Institute of Physics, Prague, Czech Republic.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808011501/fj2112sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011501/fj2112Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report