


Abstract
We have investigated the role of 2′-OH groups in the specific interaction between the acceptor stem of Escherichia coli tRNACys and cysteine-tRNA synthetase. This interaction provides for the high aminoacylation specificity observed for cysteine-tRNA synthetase. A synthetic RNA microhelix that recapitulates the sequence of the acceptor stem was used as a substrate and variants containing systematic replacement of the 2′-OH by 2′-deoxy or 2′-O-methyl groups were tested. Except for position U73, all substitutions had little effect on aminoacylation. Interestingly, the deoxy substitution at position U73 had no effect on aminoacylation, but the 2′-O-methyl substitution decreased aminoacylation by 10-fold and addition of the even bulkier 2′-O-propyl group decreased aminoacylation by another 2-fold. The lack of an effect by the deoxy substitution suggests that the hydrogen bonding potential of the 2′-OH at position U73 is unimportant for aminoacylation. The decrease in activity upon alkyl substitution suggests that the 2′-OH group instead provides a monitor of the steric environment during the RNA–synthetase interaction. The steric role was confirmed in the context of a reconstituted tRNA and is consistent with the observation that the U73 base is the single most important determinant for aminoacylation and therefore is a site that is likely to be in close contact with cysteine-tRNA synthetase. A steric role is supported by an NMR-based structural model of the acceptor stem, together with biochemical studies of a closely related microhelix. This role suggests that the U73 binding site for cysteine-tRNA synthetase is sterically optimized to accommodate a 2′-OH group in the backbone, but that the hydroxyl group itself is not involved in specific hydrogen bonding interactions.
INTRODUCTION
The RNA–protein interaction between a tRNA and a specific aminoacyl-tRNA synthetase provides the basis for aminoacylation of the tRNA. In this aminoacylation, an enzyme-bound activated amino acid is condensed with the terminal ribose in the acceptor end of a tRNA to establish the relationship between the amino acid and the tRNA anticodon triplet of the genetic code. The importance of aminoacylation emphasizes the need to understand, at the molecular and biochemical levels, the RNA–protein interaction that confers the specificity of aminoacylation.
All tRNAs fold into an L-shaped tertiary structure that consists of two coaxially stacked helices, the acceptor-TΨC helix and the dihydrouridine (D)-anticodon helix. The acceptor-TΨC helix is terminated with a CCA sequence at the 3′-end (Fig. 1), where the ribose of A76 is the site of amino acid attachment. This CCA end is distantly separated from the anticodon at the end of the D-anticodon helix by ∼75 Å. The division of tRNA into two helices coincides with the division of synthetases into two domains (1). The more ancient domain interacts with the tRNA acceptor end and contains conserved sequence and structural motifs that form the catalytic site for aminoacylation (2,3). The second domain of synthetases interacts with the tRNA anticodon and is less conserved (4,5).
Figure 1.

(A) Sequence and cloverleaf structure of E.coli tRNACys, where the boxed U73 is the major determinant for aminoacylation with cysteine. (B) Sequence and secondary structure of the synthetic E.coli microhelixCys, where U73 is boxed. (C) Sequence and secondary structure of the synthetic M.pneumoniae microhelixCys, where U73 is boxed. (D) Sequence and cloverleaf structure of B.subtilis tRNACys. The arrow indicates separation of the 5′- and 3′-fragments for reconstitution.
Studies of the RNA–protein interaction between the tRNA acceptor end and the catalytic domain of its cognate aminoacyl-tRNA synthetase are of interest because they provide insight into the historical development of the aminoacylation system. The most important aspect of aminoacylation is specificity. Of the 20 families of tRNA iso-acceptor species, at least 11 contain specific information for aminoacylation in the acceptor end of the acceptor-TΨC helix (6–9). For these 11 families of tRNAs, synthetic RNA helices that recapitulate sequences of the acceptor end are substrates for aminoacylation. Although the catalytic efficiency of RNA helices is reduced from that of their full-length tRNAs, the specificity of aminoacylation is maintained. This maintenance of specificity has been shown to be determined by nucleotides located in the first 3 bp of the acceptor stem and at the N73 nucleotide prior to the CCA end (6,10). While the specificity nucleotides vary in location, their importance in aminoacylation has been clearly demonstrated. Substitution of these nucleotides abolishes the specificity of aminoacylation, while transfer of these nucleotides to different sequence frameworks confers specificity. The dominance of these nucleotides in determining the specificity of aminoacylation suggests that they form specific interactions with their aminoacyl-tRNA synthetases. In these interactions, elements of the nucleotides, such as the bases, the ribose 2′-OH groups and the phosphodiester linkages, can all make an important contribution.
In this study we have focused on the RNA–protein interaction between the acceptor stem of E.coli tRNACys and cysteine-tRNA synthetase. We have shown that a synthetic microhelix that consists of the UCCA sequence and the 7 bp of the acceptor stem, capped by a UUCG tetraloop, is a substrate for aminoacylation (Fig. 1 ; 8,11,12). The catalytic efficiency of aminoacylation of this microhelix, defined by the steady-state kinetic parameter kcat/Km, is 1.0 s–1 M–1. Although this activity is reduced from that of the full-length tRNACys (kcat/Km = 1.0 × 106 s–1 M–1) by six orders of magnitude, the microhelix recapitulates major features of aminoacylation of the tRNA. For example, alteration of the second and third base pairs of the microhelix decreases aminoacylation by 2-fold, which is the same when the alteration is made at the second and third base pairs of the full-length tRNA (11). Most importantly, the microhelix depends on U73 for aminoacylation and this dependence is also observed in the full-length tRNA. In the microhelix, substitution of U73 eliminates aminoacylation while transfer of U73 to RNA helices of a different specificity confers on the latter the ability to accept cysteine (8). In the full-length tRNACys substitution of U73 results in a loss of 7.1 kcal/mol of free energy change of activation, which is the largest among mutations of acceptor stems that have been examined (11,13). We have recently shown that of the functional groups on the base of U73, it is the 4-carbonyl group that is responsible for the free energy change (12). Kinetic analysis has suggested that the 4-carbonyl group of U73 is an important binding site during the transition state of the RNA–synthetase interaction (12).
The primary interest here was to examine the contribution of 2′-OH groups of the microhelix to aminoacylation by cysteine-tRNA synthetase. While we have determined that the base of U73 and those of the second and third base pairs of the acceptor stem are important for aminoacylation, there is little information on the significance of 2′-OH groups. However, the importance of the 2′-OH groups in RNA structure and function has been well established. Studies of several RNA–RNA and RNA–protein interactions have shown a number of mechanisms by which a 2′-OH group contributes to RNA specificity and catalysis. The most common mechanism is through the ability of 2′-OH groups to provide hydrogen bond (H bond) donors and acceptors. The second is the ability of 2′-OH groups to influence sugar puckering. While 2′-OH groups favor the 3′-endo sugar conformation, a 2′-deoxy group favors the 2′-endo conformation (14). Recently a third mechanism has been suggested. Analysis of the crystal structure of an RNA duplex showed that 2′-OH groups propagate the stable and regular arrangement of water molecules along the major and minor grooves of the RNA helix (14), having an impact on the structure and function of the RNA. Based on these three known functions of 2′-OH groups, we investigated the role of 2′-OH groups in the interaction of the acceptor stem of E.coli tRNACys with cysteine-tRNA synthetase.
MATERIALS AND METHODS
RNA microhelices
RNA oligonucleotides and mixed RNA–DNA oligonucleotides were chemically synthesized. Sequences related to E.coli tRNACys were synthesized by the University of Pennsylvania Cancer Center Nucleic Acid Facility, while those related to Mycoplasma pneumoniae and Bacillus subtilis tRNAs were synthesized by Dharmacon Research (Boulder, CO) or by the University of Utah DNA/peptide synthesis facility. The 2′-O-propyl uridine phosphoramidite was kindly provided by Isis Pharmaceuticals and prepared as described (15). All oligonucleotides were purified by electrophoresis on 15% polyacrylamide gels containing 7 M urea, visualized by UV, eluted from gels and desalted using C18 reverse phase columns (8). The purity of oligonucleotides was confirmed by HPLC analysis. Additionally, the composition of the 2′-O-propyl-containing RNAs was verified by MALDI/MS analysis at the University of Utah. The all ribose microhelices of the E.coli and M.pneumoniae sequences were tested for plateau levels of aminoacylation by substrate levels of cysteine-tRNA synthetase. All helices showed 80–85% plateau and their concentrations were adjusted accordingly.
Aminoacylation with cysteine
Aminoacylation with cysteine of RNA helices was determined under previously established steady-state conditions (8). The wild-type all ribose E.coli helix was assayed in the range 22.5–200 µM with 2 µM cysteine-tRNA synthetase to determine the kinetic parameters kcat, Km and kcat/Km. All variant helices were assayed at 40 µM, which was one-quarter of the Km of the wild-type E.coli helix. Under this condition the initial rate of aminoacylation of the variants was taken as an estimation of the kcat/Km value.
Synthesis and purification of NMR samples
Large-scale synthesis of RNA oligonucleotides for NMR was prepared on the 1 or 10 µmol scale with 0.05 M acetonitrile solutions of PAC-protected phosphoramidites (Glen Research). The RNA oligonucleotides were then purified by anion exchange chromatography using a Pharmacia Resource Q column (16). The eluted RNA was lyophilized and redissolved in H2O and dialyzed against 1 M NaCl, 10 mM EDTA (1 × 1 l) and then against H2O (2 × 1 l). The RNA was lyophilized and dissolved in NMR buffer containing 10 mM NaPO4, 50 mM NaCl and 50 mM KCl pH 6.5, or 10 mM NaPO4 without additional monovalent salts. The final concentration of RNA for the NMR analysis was 1.5–2.5 mM.
NMR data acquisition and processing
NMR data were collected on a Varian Inova 600 MHz or Varian Unity 500 MHz NMR spectrometer. The 1D imino proton spectra were obtained at temperatures between 5 and 55°C using 1-1 solvent suppression (17), while the 2D NOESY spectra were obtained in 90% H2O/10% D2O and 99.9% D2O with mixing times between 50 and 400 ms. The 2D NOESY spectra in 90% H2O buffer were collected at 15 or 25°C with mixing times between 50 and 400 ms using a NOESY pulse sequence with a flip-back WATERGATE element (18). The 2D NOESY spectra in 99.9% D2O were acquired at temperatures of 15, 25 and 35°C with mixing times of 50, 100, 150, 250 and 400 ms. TOCSY (19) and DQCOSY (20) NMR experiments were used to help make sequential assignments and to determine sugar conformations based on coupling constant measurements (21). Data was processed and analyzed with VNMR (Varian) and Felix (MSI) software. The exchangeable and non-exchangeable base protons and sugar H1′, H2′ and H3′ protons were completely assigned, with the exception of the H3′ proton of A66. Partial resonance assignments were made for the H4′, H5′ and H5′′ protons, but resonance overlap in the sugar proton region of the homonuclear 2D NMR spectra precluded complete, unambiguous assignments.
Structure refinement
Three-dimensional structures of the M.pneumoniae tRNACys microhelix were determined from NOESY and DQCOSY NMR experiments. The structure of the central 4 bp (U3-C6 and G67-A70) was restrained to an idealized A-form helix based on NOESY connectivities and because DQCOSY data indicated that the sugars were 100% 3′-endo. For the other three base pairs (G1-C72, G2-C71 and U7-A66) in the stem, no particular conformation was enforced during the structure calculations. The base pairing was maintained using hydrogen bonding restraints set to 1.8 ± 0.1 and 2.8 ± 0.1 Å between the hydrogen heavy atom and the two heavy atoms, respectively. Restraints between U7 (O2) and A66 (H2) were used to maintain base pair planarity. The sugars for the terminal 3 bp were restricted to the 3′-endo conformation because no H1′–H2′ DQCOSY cross-peak was observed. Initial structures were generated using Insight (MSI) and refined using restrained molecular dynamics with Discover (MSI) and the AMBER force field. The UUCG tetraloop structure was determined de novo from our NMR data as a control for the quality of the structure and the final structure of this element was qualitatively the same as previously reported (22). Although the structure is at a preliminary stage due to the lack of heteronuclear, multi-dimensional NMR data, the nucleoside sugar conformations in the acceptor stem are well defined. Identification of a large number of NOEs has defined the fold-back topology and established the base stacking arrangement as shown in Figure 4.
Figure 4.

Selected regions of the 250 ms mixing time NOESY spectrum acquired at 15°C of the M.pneumoniae microhelix tRNACys. These sections show the NOE connectivities between the nucleotides of the closing stem base pair and the UCCA tail.
RESULTS
Substitution of 2′-OH groups with 2′-H in the E.coli microhelix
We began by testing the effect of substituting individual 2′-OH groups with 2′-deoxy groups in the E.coli microhelix. We prepared the all ribose wild-type microhelix by chemical synthesis and variants of the helix by incorporating a 2′-deoxy nucleoside at defined positions. In principle, the introduction of a deoxy analog would eliminate the H bond donor and acceptor ability of a 2′-OH group (Fig. 2A and B). If a particular H bond was important for aminoacylation, either to form a specific RNA structure, to interact with a residue in the synthetase or to maintain water hydration at a specific position, we expected that elimination of this H bond would decrease aminoacylation. Additionally, a deoxy analog would decrease the propensity of a sugar to adopt the 3′-endo conformation and favor the 2′-endo conformation. If the sugar conformation at a specific position was important for aminoacylation we expected that replacement by the deoxy nucleotide would have a negative effect.
Figure 2.

Chemical structures and features of the (A) 2′-OH, (B) 2′-H, (C) 2′-O-methyl and (D) 2′-O-propyl moieties on uridine.
We prepared 12 variants of the microhelix, each containing a 2′-deoxy substitution at a single position or at both positions of a base pair (Table 1). These 12 variants collectively encompassed 2′-OH groups of the first 3 bp positions, where specific nucleotides have been implicated as important for aminoacylation (11). Three of the 12 variants covered the 2′-OH groups of the single-stranded U73, C74 and C75. We did not test the 2′-OH group of the terminal A76, as this position is believed to be the site of amino acid attachment for cysteine-tRNA synthetase. Studies of aminoacyl-tRNA synthetases have shown that a subclass of these enzymes, the class I synthetases, aminoacylate at the 2′-OH of the terminal ribose, whereas the class II synthetases aminoacylate at the 3′-OH (3,5). Escherichia coli cysteine-tRNA synthetase is a member of the class I synthetases (23,24).
Table 1. Relative aminoacylation of microhelices by E.coli cysteine-tRNA synthetase.
| Position of mutation |
All ribose |
2′-Deoxy |
2′-O-methyl |
| E.coli microhelixCys (wild-type) | 1.00 | ||
| C75 | 0.90 ± 0.09 | 0.47 ± 0.06 | |
| C74 | 1.00 ± 0.09 | 0.43 ± 0.12 | |
| U73 | 2.10 ± 0.43 | 0.08 ± 0.01 | |
| G1:C72 | 0.25 ± 0.08 | 0.47 ± 0.06 | |
| G1 | 0.60 ± 0.13 | n.d. | |
| C72 | 0.45 ± 0.08 | n.d. | |
| G2:C71 | 4.86 ± 0.37 | 0.87 ± 0.21 | |
| G2 | 0.51 ± 0.04 | 0.87 ± 0.04 | |
| C71 | 10.9 ± 0.7 | 0.32 ± 0.01 | |
| C3:G70 | 2.22 ± 0.38 | 0.29 ± 0.02 | |
| C3 | 0.20 ± 0.06 | 0.44 ± 0.11 | |
| G70 | 0.95 ± 0.05 | 0.20 ± 0.08 |
The kinetic parameters of the all ribose E.coli helix were kcat = 6.0 × 10–5 s–1, Km = 60 µM and kcat/Km = 1.0 s–1 M–1. These were derived from a Lineweaver–Burk plot of the Michaelis–Menten equation based on activity determined at 22.5–200 µM RNA substrate. All other helices were assayed at 40 µM and their initial rates were taken as an approximation of kcat/Km. Individual kcat/Km values were normalized to that of the E.coli helix to obtain relative values. Each value was the average of at least two determinations. The positions of substitutions are shown as in the microhelix (Fig. 1). n.d., not determined.
We measured steady-state kinetics of aminoacylation for the wild-type (all ribose) and variant microhelices, using the kcat/Km value for the wild-type as a reference. The kcat/Km value is a measure of the catalytic efficiency of aminoacylation and is a parameter that can be converted to the free energy change of activation. Based on previously established conditions that satisfied Michaelis–Menten kinetics (8,12), we assayed the wild-type microhelix over a range of concentrations from 22.5 to 200 µM with 2 µM purified E.coli cysteine-tRNA synthetase. Analysis of the Lineweaver–Burk plot gave a Km of 60 µM and a kcat of 6.0 × 10–5 s–1 (Table 1), both of which were similar to those obtained previously (8,12). The kcat/Km value was 1.0 s–1 M–1, which was in agreement with previous values (12). We performed kinetic analysis for variants at substrate concentrations at least 4-fold below the Km of the wild-type. At these substrate concentrations the initial rate of aminoacylation was approximately proportional to kcat/Km.
Of the 12 deoxy variants none showed a major decrease in kcat/Km from that of the wild-type (Table 1). Specifically, the three nucleotides in the single-stranded region, U73, C74 and C75, were insensitive to the substitution. The variants containing deoxy at C74 and C75 essentially maintained the activity of the wild-type, while that containing deoxy at U73 had a slightly increased activity (by 2-fold). At the G1:C72 position the two nucleotides showed a minor sensitivity to the deoxy substitution. Single substitution of the G1 and C72 nucleotides decreased the relative activity to 0.60 and 0.45, respectively, while double substitution of G1:C72 decreased the activity to 0.25. Because the decrease in activity of the double substitution appeared to be the product of the single substitutions, the effect of the single substitutions appeared to be independent and additive. At the G2:C71 position the two nucleotides also showed little sensitivity, although they responded differently to substitution. While substitution of the G2 nucleotide decreased the relative activity to 0.51, that of C71 increased the activity to 10.9. The double substitution showed an activity increase to 4.86. Thus, there was a small deleterious effect for the deoxy substitution at position G2, but an advantageous effect at position C71. The effect of the double substitution appeared to be the product of the two single substitutions. At the C3:G70 position substitution at the C3 position decreased activity to 0.20, but that of G70 had no effect. The double substitution showed an increase in activity to 2.22.
Overall, there was no major defect caused by introduction of the deoxynucleotide substitution. The decrease in activity was minor compared to the decrease by orders of magnitude seen for substitution of functional groups on the base of U73 (12). To confirm the minor effect of the deoxy substitutions we tested the variant that contained 2′-deoxy at all but the A76 position. This variant showed a relative activity of 0.27, which was only 4-fold lower than that of the all ribose sequence (Table 2). Thus, even the microhelix containing a deoxy backbone (except for the terminal residue) did not suffer a major decrease in aminoacylation. Because deoxynucleosides cannot act as H bond donors/acceptors, this suggests that potential H bonds involving the 2′-OH groups in the microhelix are not important for aminoacylation. Deoxy substitution at several positions actually increased aminoacylation, which could be due to elimination of intramolecular H bonds that constrain the local sugar conformation or the overall flexibility of the RNA during aminoacylation.
Table 2. Relative aminoacylation of microhelices by E.coli cysteine-tRNA synthetase.
| Substrate | Relative kcat/Km |
|
| |
E.coli microhelixCys |
M.pneumoniae helixCys |
| All ribose helix | 1.0 | 0.86 ± 0.14 |
| All deoxy helix except for A76 | 0.27 ± 0.05 | 0.47 ± 0.02 |
| Deoxy U73 | 2.10 ± 0.43 | 1.78 ± 0.05 |
| 2′-O-methyl U73 | 0.08 ± 0.01 | 0.10 ± 0.01 |
| 2′-O-propyl U73 | 0.038 ± 0.005 | 0.037 ± 0.005 |
The E.coli helix has a kcat of 6.0 × 10–5 s–1, a Km of 60 µM and a kcat/Km of 1.0 s–1 M–1. These kinetic parameters were determined under the conditions described in the legend to Table 1. The other helices were assayed at 40 µM and their initial rates were taken as an approximation of the kcat/Km values. Individual kcat/Km values were normalized to that of the E.coli helix to obtain the relative values. Each value was the average of at least two determinations.
Substitution of 2′-OH groups with 2′-O-methyl groups in the E.coli microhelix
We prepared a second set of mutants by introducing 2′-O-methyl groups to replace 2′-OH groups. Compared to the 2′-deoxy substitution, this replacement serves the dual purpose of maintaining the hydrogen acceptor activity and the 3′-endo sugar conformational preference of the normal ribose (Fig. 2C). We examined 10 variants containing a 2′-O-methyl substitution, which encompassed positions of the first 3 bp and of the single-stranded 73–75 nt. In general, the 2′-O-methyl substitution decreased aminoacylation, but the decrease was minor and there was no increase in activity by substitution at any of the positions tested (Table 1). Nine of the 10 variants had 2- to 5-fold decreased activity compared to that of the wild-type. Some of these variants, such as mutants containing a substitution at positions C75, C74, G1:C72, G2, C3 and G70, showed a similar degree of decrease as that of their 2′-deoxy counterparts. The lack of sensitivity to substitution with either a deoxy or 2′-O-methyl group clearly indicated that the H bonding potential of the 2′-OH groups was not critical for aminoacylation nor was the 3′-endo sugar conformation important. Other variants, such as those containing a substitution at positions G2:C71, C71 and C3:G70, showed a decrease in aminoacylation for the 2′-O-methyl substitution. However, their 2′-deoxy counterparts showed an increase in aminoacylation. Although the molecular basis for the decrease in aminoacylation by the 2′-O-methyl substitution is not clear, the degree of decrease for these variants was minimal.
Only one of the 10 variants containing a 2′-O-methyl substitution showed as much as a 10-fold decrease in activity. The substitution in this variant was at position U73, which is the major determinant for aminoacylation with cysteine. The 2′-O-methyl substitution at U73 reduced the activity to 0.08 relative to that of the wild-type, which was a significant decrease compared to all other substitutions. This decrease indicated that introduction of the 2′-O-methyl group interfered with aminoacylation by cysteine-tRNA synthetase. The basis for the decrease cannot be due to the H bond accepting property or the 3′-endo sugar conformation of the 2′-O-methyl group, as these features are common with the normal 2′-OH group. Additionally, the decrease cannot be due to the lack of a H bond donor for the 2′-O-methyl group, as this feature is also absent from the 2′-deoxy substituted RNA (Table 1). Taken together, these considerations argue against a role of H bonds or sugar conformation of the 2′-OH of U73 in aminoacylation with cysteine. Instead, they suggest that the basis for the decrease in activity by the 2′-O-methyl substitution is due to the bulkier size of the methyl group.
Substitution of 2′-OH groups in the M.pneumoniae microhelix
We chose the acceptor stem of M.pneumoniae tRNACys as a second sequence framework to examine the role of 2′-OH groups in aminoacylation. The purpose was to determine if general features of 2′-OH groups of the E.coli helix could be reproduced in a different sequence. We have recently presented a preliminary NMR model of the M.pneumoniae tRNACys acceptor stem (25). This model has helped to establish a structural hypothesis from which the role of 2′-OH groups could be interpreted. We synthesized the M.pneumoniae microhelix containing the 7 bp of the acceptor stem of the tRNA together with the UCCA terminal sequence and the UUCG tetraloop to cap the acceptor stem of the microhelix (Fig. 1C). This microhelix shared in common with the E.coli sequence the U73 nucleotide, the first two base pairs and the fourth and fifth base pairs in the acceptor stem.
We replaced 2′-OH groups in the M.pneumoniae microhelix by either a 2′-deoxy or 2′-O-ethyl substitution and determined the effect of these substitutions on aminoacylation by E.coli cysteine-tRNA synthetase. The E.coli enzyme was used as a reference so that we could compare the effect on the M.pneumoniae helix with that on the E.coli helix. The E.coli enzyme is able to aminoacylate microhelices of different sequences, provided that U73 is present (8). As expected, the E.coli enzyme aminoacylated the wild-type M.pneumoniae microhelix with a kcat/Km of 0.86 relative to that of the E.coli helix (Table 2). This confirmed that the M.pneumoniae helix was a substrate comparable to the E.coli helix.
In general, the effect of substitution of 2′-OH groups in the M.pneumoniae helix was parallel to that in the E.coli helix (Table 2). The variant of the M.pneumoniae helix containing all but A76 as 2′-deoxy had a relative kcat/Km of 0.47. This was similar to the relative kcat/Km of 0.27 of its E.coli counterpart. The small effect further emphasized that the absence of H bonding potential by the deoxy substitution was not critical for aminoacylation. At the position of the major determinant U73 the single deoxy substitution caused a small increase of 1.78-fold in kcat/Km, which was reminiscent of the 2.10-fold increase in kcat/Km in the E.coli counterpart. The single 2′-O-methyl substitution decreased kcat/Km by 10-fold, which reproduced the effect of the substitution in the E.coli helix. These results are all consistent with a model in which the 2′-OH group of U73 is tolerant of a smaller group, such as the deoxy group, but not tolerant of a larger group, such as 2′-O-methyl.
To test the model further we introduced the even larger 2′-O-propyl group to the U73 position of the E.coli and M.pneumoniae microhelices. The propyl group is larger than the methyl group by two methylenes (Fig. 2D) and was predicted to further decrease the aminoacylation activity. We synthesized variants of both the E.coli and M.pneumoniae microhelices containing a 2′-O-propyl group at position U73 and tested their ability to be aminoacylated by E.coli cysteine-tRNA synthetase. As expected, the activity of both helices was decreased to ∼0.04 relative to that of the E.coli wild-type helix. This decrease was more severe than the 10-fold decrease seen for the 2′-O-methyl substitution.
Substitution of the 2′-OH group of U73 in a reconstituted tRNA
The significance of the 2′-OH group of U73 was tested in a reconstituted tRNA to examine the effect of substitution in the context of the whole tRNA molecule. Functional reconstitution of several tRNAs has been achieved by annealing a T7 transcribed 5′-fragment with a chemically synthesized 3′-fragment to give rise to a tRNA containing just one break (26,27). A convenient break point is at position 57 using a DNA template cleaved at the TaqI restriction site in the T loop (Fig. 1). However, E.coli tRNACys was not suitable for this strategy as reconstitution decreased the activity by >103-fold. Possibly this is because the break point is within the tRNA tertiary core, which may be harder to reconstitute in E.coli tRNACys which has an unusual G15:G48 base pair (28). We therefore used B.subtilis tRNACys as substrate, because it contains the normal G15:C48 and is efficiently aminoacylated by E.coli cysteine-tRNA synthetase (Table 3 and Fig. 1D) (29). As expected, the reconstituted B.subtilis tRNACys was an active substrate, albeit with a 50-fold reduced kcat/Km relative to that of the full-length E.coli tRNA (Table 3).
Table 3. Aminoacylation of reconstituted tRNACys of B.subtilis by E.coli cysteine-tRNA synthetase.
| Substrate |
Km (µM) |
kcat (s–1) |
kcat/Km (s–1 M–1) |
Relative value |
| Full-length E.coli tRNACys | 1.27 | 1.28 | 1.0 × 106 | 50 |
| Full-length B.subtilis tRNACys | 1.70 | 2.76 | 1.6 × 106 | 80 |
| U73 (ribose) in reconstituted tRNACys | 43.7 | 0.82 | 1.8 × 104 | 1.0 |
| U73 (deoxy) in reconstituted tRNACys | 38.5 | 0.64 | 1.7 × 104 | 1.0 |
| U73 (2′-O-methyl) in reconstituted tRNACys | 36.9 | 0.0063 | 1.7 × 102 | 0.01 |
Full-length tRNAs were synthesized by T7 transcription. The reconstituted tRNAs were prepared by annealing the 5′-fragment (synthesized by T7 transcription) and the 3′-fragment (synthesized chemically), containing U73 with a ribose, deoxy or 2′-O-methyl backbone. Aminoacylation was achieved with substrate concentrations in the range 0.5–8 µM for full-length transcripts, 2–16 µM for reconstituted tRNAs containing U73 with a ribose and deoxy backbone and 10–100 µM for the reconstituted tRNA containing U73 with a 2′-O-methyl backbone. The concentration of enzyme was 1 µM for the 2′-O-methyl-containing tRNA and 10 nM for all others.
Using the reconstituted B.subtilis tRNACys as a reference, we compared the activity of variants containing the deoxy and 2′-O-methyl substitutions at U73. The deoxy substitution virtually maintained the activity of the all ribose wild-type, which is in agreement with only a small effect observed in the microhelix. However, the 2′-O-methyl substitution reduced activity by 100-fold, which is 10-fold more than that observed in the microhelix. Thus, the enzyme sensitivity to the bulkier 2′-O-methyl substitution is the same whether the substitution is in a microhelix or a reconstituted tRNA. However, the degree of sensitivity is amplified in the reconstituted tRNA, which may be due to the presence of a break point in the tRNA tertiary core or to other parts of tRNA that work cooperatively with the ribose of U73.
NMR structure of an acceptor stem–loop
To gain insights into the role of the 2′-OH group of U73, NMR spectroscopy was used to determine the structure of the microhelix of M.pneumoniae tRNACys (Fig. 1C). Resonance assignments were straightforward for the stem region of the microhelix (nt 1–7 and 66–72) since intra- and internucleotide NOES typical of A-form helical RNA were observed in the 2D NOESY spectra (Fig. 4). Assignments for base paired imino protons were made by sequential connectivity between imino protons on adjacent base pairs in the stem (Fig. 3). The two U-A (U3-A70 and U7-A66) base pairs were identified by characteristic chemical shifts observed for uridine imino and adenosine H2 protons. Amino protons of the five base paired cytosines (C5, C6, C69, C71 and C72) were assigned by the NOEs to their own H5 protons and the imino proton of the guanosine with which it is base paired. NOE cross-peaks were observed between the H2 protons of the Watson–Crick base paired adenosine at position i to the H1′ proton of the next sequential (i + 1) nucleotide, as well as H1′ of the nucleotide that is base paired with nucleotide i – 1 (Fig. 4). The H2–H1′ NOEs were of nearly equal intensities, as is typical of an A-form, helical RNA. The ribose sugars in the helical region adopted primarily a 3′-endo conformation, as indicated by the absence of H1′–H2′ coupling constants in the DQCOSY spectra. Taken together, these data clearly showed that the seven base pairs in the stem adopt a canonical A-form helical conformation.
Figure 3.

Imino spectrum at 15°C of the M.pneumoniae microhelix tRNACys. The assignments are indicated above their corresponding resonance position. The imino proton resonances for U8 and G11 confirm that a UUCG tetraloop is formed. The presence of an imino peak for G1 is exceptional at this high a temperature and supports the structural model wherein stacking of A76 onto G1 serves to protect the G1 imino proton from exchange.
The tetraloop UUCG capping the microhelix is one of the most well characterized RNA structures. Previously reported proton resonance assignments (21) for this tetraloop were used as references for assignments. Characteristic resonances from the 2′-OH proton of U8 at 6.56 p.p.m. and the imino proton of G11 at 9.65 p.p.m. were observed, which are similar to the expected chemical shifts of 6.82 and 9.96 p.p.m., respectively (30). The very intense NOE cross-peak between the H8 and H1′ protons of a guanosine in the syn-conformation was observed for G11. The sugar conformations for nucleotides U9 and C10 were C2′-endo, which has been previously reported (21). These resonance assignments and sugar conformations confirmed correct formation of the UUCG tetraloop and the refined structure of the tetraloop region was qualitatively similar to that reported previously (22,30).
The UCCA tail of the microhelix adopts a fold-back structure that places nucleotide A76 in close proximity to G1. This feature has been qualitatively described in an NMR analysis of a mutant tRNAfMet (31). We observed NOEs between nucleotides C72-U73 and U73-C74, which indicate base stacking between these nucleotides (Fig. 5). A total of five and nine internucleotide NOEs were observed between U73-C72 and C74-U73, respectively. A break in base stacking occurs between nucleotides C74 and C75, where only two NOEs between these residues were observed. The H6 of C75 has a weak and a medium strength NOE to the H2′ and H3′ protons of C74, respectively. If C75 was stacked in an A-form helical type structure, then a large NOE would be expected to arise between the C75 H6 and C74 H2′ protons. There are five NOEs observed between nucleotides A76 and C75, with four arising from the A76(H8)-C75(H6, H1′, H2′ and H3′) protons. These NOEs would suggest that stacking interactions occur between A76 and C75, although the stacking is not likely to be A-form in nature due to an atypical NOE arising from the C75(H6)-A76(H2′) protons. The fold-back structure of the UCCA tail is further confirmed through the observation of two weak NOEs that were previously reported between A76(H2)-G1(H1′) and A76(H1′)-G1(H1′) protons (31). In addition to these previously reported NOEs, six new weak NOEs arising from G1(H8)-A76(H8, H2, H1′ and H4′), G1(imino)-A76(H2) and G1(H1′)-A76(H8) were observed. These NOEs confirm that A76 is in close proximity to G1. Many of these NOEs would not be seen if the tail formed an A-form helical stack. The DQCOSY spectra obtained at 25°C indicate that the sugar conformations of the UCCA tail nucleotides are predominately C2′-endo.
Figure 5.
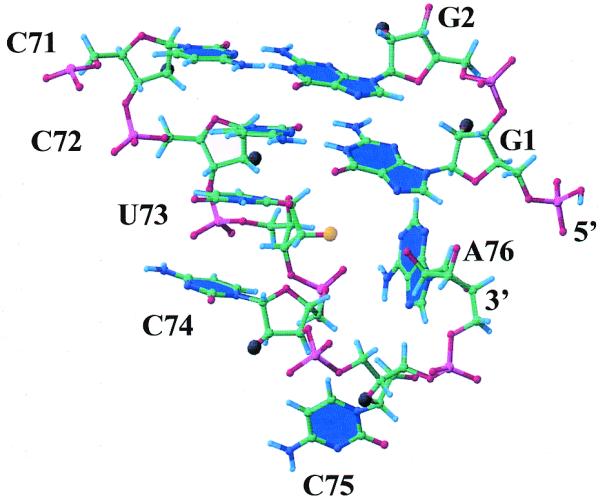
The NMR-based fold-back model of the UCCA end in the M.pneumoniae microhelixCys, which shows the bases of C71, C72, U73, C74, C75, A76, G1 and G2 as planes and 2′-OH groups as large balls. The 2′-OH group of U73 is colored orange whereas those of all others are in black.
DISCUSSION
Systematic substitution of ribose in RNA microhelices has yielded two major conclusions. First, the majority of the 2′-OH groups in these helices are unimportant for aminoacylation by cysteine-tRNA synthetase. Even elimination of all but the 2′-OH of A76 had only a minor effect. Presumably the 2′-OH groups in these helices form specific intramolecular H bonds, affect hydration and influence the sugar puckering preference of the RNA. However, none of these effects is important for aminoacylation. Second, the only 2′-OH that has a clear role in aminoacylation is on U73, the site of the major determinant for aminoacylation with cysteine. The importance of the 2′-OH group of U73 is not in its H bonding potential but in creating an appropriate steric environment for cysteine-tRNA synthetase to recognize the RNA helix. This role has been confirmed in the context of a reconstituted tRNA. While deoxy substitution of the 2′-OH of U73 does not decrease aminoacylation, substitution with bulkier analogs, such as 2′-O-methyl or 2′-O-propyl, decreases aminoacylation and the level of decrease is correlated with the size of the analog. This suggests that the 2′-OH on U73 is a steric sensor of the RNA–synthetase interaction. If the steric size at the 2′-OH position is not appropriate, cysteine-tRNA synthetase can reject the RNA.
An NMR-based model of the acceptor stem of M.pneumoniae provides a basis to understand how the 2′-OH group of U73 could be positioned to affect the RNA–synthetase interaction. This model shows a fold-back structure for the UCCA end, where the terminal A76 of the UCCA tail is not extended but is folded back to be adjacent to G1 in the acceptor stem (Fig. 5). In the fold-back structure of the UCCA tail the continuous stacking between C72, U73 and C74 creates an RNA ‘A-like’ structural environment for U73 with distinct major and minor groove sides. The 4-carbonyl group of U73, which is the most critical functional group for aminoacylation, is located on the major groove side of the acceptor stem. In contrast, the 2′-OH group of U73 is in the minor groove side, facing the interior of the fold-back loop. In this position the 2′-OH group points towards the 2′-OH group of A76, which is the site of amino acid attachment for the class I cysteine-tRNA synthetase.
Similar fold-back features of the UCCA end were also identified in an earlier NMR study of the acceptor stem of a variant of E.coli tRNAfMet (31). This study showed that while the UCCA end has a fold-back structure, the ACCA end in the same sequence context has an extended structure. Thus, the fold-back structure is dependent on U73. Substitution of U73 with A73 eliminated the fold-back and extended the CCA end from the acceptor stem. In the extended conformation the 2′-OH at position 73 is separated from the 2′-OH of A76 and would not be expected to influence aminoacylation. The dependence of the fold-back on U73 is also supported by biochemical studies (32,33). Furthermore, we have used T4 RNA ligase to catalyze joining of the tRNA 3′- and 5′-ends and used the rate of ligation to estimate the proximity of the two ends. The wild-type E.coli tRNACys, containing the UCCA end, is a better substrate for the joining reaction than the A73 variant (34). The faster joining of the UCCA end is consistent with the fold-back structure that brings the terminal A76 to the vicinity of G1. The slower joining of the A73 variant is consistent with the observed extended conformation that separates A76 from G1.
Thus, as a result of fold-back of the UCCA end, the 2′-OH of U73 is positioned between the two functional groups that are most important for aminoacylation. One is the 4-carbonyl group of U73, which is the major determinant for recognition by cysteine-tRNA synthetase, and the other is the 2′-OH of A76, which is the site of aminoacylation. During the transition state of the reaction the enzyme must contact the 4-carbonyl group of U73 while catalyzing the aminoacylation reaction at the 2′-OH of A76. The delicate location of the 2′-OH of U73, between the 4-carbonyl of U73 and 2′-OH of A76, provides a rationale for a close physical contact at this site with cysteine-tRNA synthetase during the process of aminoacylation. As the group becomes larger, from 2′-OH to 2′-O-methyl to 2′-O-propyl, the introduction of steric hindrance would interfere with aminoacylation. On the other hand, a reduction in size would have little or no effect, as long as the conformation is not significantly changed. Overall, biochemical studies and the NMR model of the RNA structure complement each other and both suggest strategic placement of a 2′-OH group.
Enzymatic discrimination based on the steric bulk of U73 has not been documented previously. Studies of other RNA–protein and RNA–RNA interactions have identified important 2′-OH groups largely on the basis of H bond formation. The significance of H bonds is interpreted from loss of activity due to substitution with a 2′-deoxy group. Such a loss of activity has been observed for 2′-OH groups that cluster around the G3:U70 major determinant of tRNAAla for aminoacylation by alanine-tRNA synthetase (35). Loss of activity has also been observed on substitution of a 2′-OH group that forms an H bond within the core region of tRNAPro (36). This H bond is critical for maintenance of the tRNA structure and for aminoacylation by proline-tRNA synthetase. Also, a 2′-OH group in the RNA operator of the MS2 coat protein has been identified as important (37). Structural analysis of this RNA–protein complex suggests that the 2′-OH forms a critical H bond between the RNA and the coat protein (37,38). Further, several 2′-OH groups in the polypyrimidine tract adjacent to 3′ splice sites of eukaryotic mRNAs are important for the specific splicing repressor Sex lethal protein (39). These 2′-OH groups, however, are not recognized by the general splicing factor U2AF65 and as such they provide a discriminatory mechanism that differentiates the specific splicing repressor from the general splicing factor. In RNA catalysis by the group I and group II introns (40,41), the hammerhead ribozyme (42) and the P RNA of RNase P (43) important 2′-OH groups have been identified that form H bonds within the catalytic center or with the RNA substrate.
The discovery of a discriminatory function of a 2′-OH group in the acceptor end of a tRNA suggests an ancient history of this function. Because aminoacylation of RNA helices is believed to be one of the earliest steps in the evolution of the present day decoding machinery (44,45), identification of this function has historical significance. In development of the L-shaped tRNA structure some 2′-OH groups located outside the acceptor end might have gained importance. Recently, an examination of the 2′-OH groups in the full-length E.coli tRNAAla has revealed new sites that are important for aminoacylation in addition to those previously identified in the acceptor stem (46). These new sites suggest that additional 2′-OH groups have been selected to enhance aminoacylation of the full-length tRNA. Nonetheless, studies of 2′-OH groups in the acceptor end that are important for aminoacylation can provide insights into the primordial decoding system. With the growing database of RNA structures, due in large part to recent high resolution structures of the bacterial ribosome (47–49), additional examples of the discriminatory function of a 2′-OH group may be identified to shed more light on the ancient ‘RNA world’.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the University of Pennsylvania Cancer Center Nucleic Acid Facility for providing special services. This work was supported by grants from the National Institutes of Health GM56662 to Y.M.H. and GM/AI55508 to D.R.D.
References
- 1.Schimmel P., Giege,R., Moras,D. and Yokoyama,S. (1993) An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl Acad. Sci. USA, 90, 8763–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moras D. (1993) Structural aspects and evolutionary implications of the recognition between tRNAs and aminoacyl-tRNA synthetases. Biochimie, 75, 651–657. [DOI] [PubMed] [Google Scholar]
- 3.Cusack S. (1997) Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol., 7, 881–889. [DOI] [PubMed] [Google Scholar]
- 4.Carter C.W.,Jr (1993) Cognition, mechanism and evolutionary relationships in aminoacyl-tRNA synthetases. Annu. Rev. Biochem., 62, 715–748. [DOI] [PubMed] [Google Scholar]
- 5.Arnez J.G. and Moras,D. (1997) Structural and functional considerations of the aminoacylation reaction. Trends Biochem. Sci., 22, 211–216. [DOI] [PubMed] [Google Scholar]
- 6.Martinis S.A. and Schimmel,P. (1995) Small RNA oligonucleotide substrates for specific aminoacylations. In Soll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. American Society of Microbiology Press, Washington, DC, pp. 349–370.
- 7.Frugier M., Florentz,C. and Giege,R. (1994) Efficient aminoacylation of resected RNA helices by class II aspartyl-tRNA synthetase dependent on a single nucleotide. EMBO J., 13, 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamann C.S. and Hou,Y.M. (1995) Enzymatic aminoacylation of tRNA acceptor stem helices with cysteine is dependent on a single nucleotide. Biochemistry, 34, 6527–6532. [DOI] [PubMed] [Google Scholar]
- 9.Quinn C.L., Tao,N. and Schimmel,P. (1995) Species-specific microhelix aminoacylation by a eukaryotic pathogen tRNA synthetase dependent on a single base pair. Biochemistry, 34, 12489–12495. [DOI] [PubMed] [Google Scholar]
- 10.Francklyn C., Shi,J.P. and Schimmel,P. (1992) Overlapping nucleotide determinants for specific aminoacylation of RNA microhelices. Science, 255, 1121–1125. [DOI] [PubMed] [Google Scholar]
- 11.Hou Y.M., Sterner,T. and Bhalla,R. (1995) Evidence for a conserved relationship between an acceptor stem and a tRNA for aminoacylation. RNA, 1, 707–713. [PMC free article] [PubMed] [Google Scholar]
- 12.Hou Y.M., Sundaram,M., Zhang,X., Holland,J.A. and Davis,D.R. (2000) Recognition of functional groups in an RNA helix by a class I tRNA synthetase. RNA, 6, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komatsoulis G.A. and Abelson,J. (1993) Recognition of tRNA(Cys) by Escherichia coli cysteinyl-tRNA synthetase. [Erratum published in Biochemistry (1993), 32, 13374.] Biochemistry, 32, 7435–7444. [DOI] [PubMed] [Google Scholar]
- 14.Egli M., Portmann,S. and Usman,N. (1996) RNA hydration: a detailed look. Biochemistry, 35, 8489–8494. [DOI] [PubMed] [Google Scholar]
- 15.Lesnik E.A., Guinosso,C.J., Kawasaki,A.M., Sasmor,H., Zounes,M., Cummins,L.L., Ecker,D.J., Cook,P.D. and Freier,S.M. (1993) Oligodeoxynucleotides containing 2′-O-modified adenosine: synthesis and effects on stability of DNA:RNA duplexes. Biochemistry, 32, 7832–7838. [DOI] [PubMed] [Google Scholar]
- 16.Wincott F., DiRenzo,A., Shaffer,C., Grimm,S., Tracz,D., Workman,C., Sweedler,D., Gonzalez,C., Scaringe,S. and Usman,N. (1995) Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res., 23, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plateau P. and Gueron,M. (1982) Exchangeable proton NMR without base-line distortion. Using new strong-pulse sequences. J. Am. Chem. Soc., 104, 7310–7311. [Google Scholar]
- 18.Mori S., Abeygunawardana,C., Johnson,M.O. and van Zijl,P.C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. [Erratum published in J. Magn. Reson. B (1996), 110, 321.] J. Magn. Reson. B, 108, 94–98. [DOI] [PubMed] [Google Scholar]
- 19.Levitt M., Freeman,R. and Frenkel,T. (1982) Broadband heteronuclear decoupling. J. Magn. Reson., 47, 328–330. [Google Scholar]
- 20.Piantini U., Sorenson,A.W. and Ernst,R.R. (1982) Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc., 104, 6800–6801. [Google Scholar]
- 21.Varani G., Cheong,C. and Tinoco,I.,Jr (1991) Structure of an unusually stable RNA hairpin. Biochemistry, 30, 3280–3289. [DOI] [PubMed] [Google Scholar]
- 22.Cheong C., Varani,G. and Tinoco,I.,Jr (1990) Solution structure of an unusually stable RNA hairpin, 5′GGAC(UUCG)GUCC. Nature, 346, 680–682. [DOI] [PubMed] [Google Scholar]
- 23.Hou Y.M., Shiba,K., Mottes,C. and Schimmel,P. (1991) Sequence determination and modeling of structural motifs for the smallest monomeric aminoacyl-tRNA synthetase. Proc. Natl Acad. Sci. USA, 88, 976–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eriani G., Dirheimer,G. and Gangloff,J. (1991) Cysteinyl-tRNA synthetase: determination of the last E. coli aminoacyl- tRNA synthetase primary structure. Nucleic Acids Res., 19, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland J.A., Hou,Y.-M. and Davis,D.R. (1999) NMR structural studies of the tRNACys amino acceptor stem of Mycoplasma pneumonia. Nucleic Acids Symp. Ser., 41, 101–103. [Google Scholar]
- 26.Yap L.P., Stehlin,C. and Musier-Forsyth,K. (1995) Use of semi-synthetic transfer RNAs to probe molecular recognition by Escherichia coli proline-tRNA synthetase. Chem. Biol., 2, 661–666. [DOI] [PubMed] [Google Scholar]
- 27.Hou Y.M. (2000) Unusual synthesis by the Escherichia coli CCA-adding enzyme. RNA, 6, 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christian T., Lipman,R.S., Evilia,C. and Hou,Y.M. (2000) Alternative design of a tRNA core for aminoacylation. J. Mol. Biol., 303, 503–514. [DOI] [PubMed] [Google Scholar]
- 29.Hou Y.M., Motegi,H., Lipman,R.S., Hamann,C.S. and Shiba,K. (1999) Conservation of a tRNA core for aminoacylation. Nucleic Acids Res., 27, 4743–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allain F.H. and Varani,G. (1995) Structure of the P1 helix from group I self-splicing introns. J. Mol. Biol., 250, 333–353. [DOI] [PubMed] [Google Scholar]
- 31.Puglisi E.V., Puglisi,J.D., Williamson,J.R. and RajBhandary,U.L. (1994) NMR analysis of tRNA acceptor stem microhelices: discriminator base change affects tRNA conformation at the 3′ end. Proc. Natl Acad. Sci. USA, 91, 11467–11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee C.P., Dyson,M.R., Mandal,N., Varshney,U., Bahramian,B. and RajBhandary,U.L. (1992) Striking effects of coupling mutations in the acceptor stem on recognition of tRNAs by Escherichia coli Met-tRNA synthetase and Met-tRNA transformylase. Proc. Natl Acad. Sci. USA, 89, 9262–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C.P., Mandal,N., Dyson,M.R. and RajBhandary,U.L. (1993) The discriminator base influences tRNA structure at the end of the acceptor stem and possibly its interaction with proteins. Proc. Natl Acad. Sci. USA, 90, 7149–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou Y.M., Lipman,R.S. and Zarutskie,J.A. (1998) A tRNA circularization assay: evidence for the variation of the conformation of the CCA end. RNA, 4, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musier-Forsyth K. and Schimmel,P. (1992) Functional contacts of a transfer RNA synthetase with 2′-hydroxyl groups in the RNA minor groove. Nature, 357, 513–515. [DOI] [PubMed] [Google Scholar]
- 36.Yap L.P. and Musier-Forsyth,K. (1995) Transfer RNA aminoacylation: identification of a critical ribose 2′-hydroxyl-base interaction. RNA, 1, 418–424. [PMC free article] [PubMed] [Google Scholar]
- 37.Baidya N. and Uhlenbeck,O.C. (1995) The role of 2′-hydroxyl groups in an RNA–protein interaction. Biochemistry, 34, 12363–12368. [DOI] [PubMed] [Google Scholar]
- 38.Valegard K., Murray,J.B., Stockley,P.G., Stonehouse,N.J. and Liljas,L. (1994) Crystal structure of an RNA bacteriophage coat protein–operator complex. Nature, 371, 623–626. [DOI] [PubMed] [Google Scholar]
- 39.Singh R., Banerjee,H. and Green,M.R. (2000) Differential recognition of the polypyrimidine-tract by the general splicing factor U2AF65 and the splicing repressor sex-lethal. RNA, 6, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pyle A.M. and Cech,T.R. (1991) Ribozyme recognition of RNA by tertiary interactions with specific ribose 2′-OH groups. Nature, 350, 628–631. [DOI] [PubMed] [Google Scholar]
- 41.Abramovitz D.L., Friedman,R.A. and Pyle,A.M. (1996) Catalytic role of 2′-hydroxyl groups within a group II intron active site. Science, 271, 1410–1413. [DOI] [PubMed] [Google Scholar]
- 42.Williams D.M., Pieken,W.A. and Eckstein,F. (1992) Function of specific 2′-hydroxyl groups of guanosines in a hammerhead ribozyme probed by 2′ modifications. Proc. Natl Acad. Sci. USA, 89, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan T., Loria,A. and Zhong,K. (1995) Probing of tertiary interactions in RNA: 2′-hydroxyl-base contacts between the RNase P RNA and pre-tRNA. Proc. Natl Acad. Sci. USA, 92, 12510–12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schimmel P. and Henderson,B. (1994) Possible role of aminoacyl-RNA complexes in noncoded peptide synthesis and origin of coded synthesis. Proc. Natl Acad. Sci. USA, 91, 11283–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henderson B.S. and Schimmel,P. (1997) RNA–RNA interactions between oligonucleotide substrates for aminoacylation. Bioorg. Med. Chem., 5, 1071–1079. [DOI] [PubMed] [Google Scholar]
- 46.Pleiss J.A., Wolfson,A.D. and Uhlenbeck,O.C. (2000) Mapping contacts between Escherichia coli alanyl tRNA synthetase and 2′ hydroxyls using a complete tRNA molecule. Biochemistry, 39, 8250–8258. [DOI] [PubMed] [Google Scholar]
- 47.Clemons W.M.,Jr, May,J.L., Wimberly,B.T., McCutcheon,J.P., Capel,M.S. and Ramakrishnan,V. (1999) Structure of a bacterial 30S ribosomal subunit at 5.5 Å resolution. Nature, 400, 833–840. [DOI] [PubMed] [Google Scholar]
- 48.Cate J.H., Yusupov,M.M., Yusupova,G.Z., Earnest,T.N. and Noller,H.F. (1999) X-ray crystal structures of 70S ribosome functional complexes. Science, 285, 2095–2104. [DOI] [PubMed] [Google Scholar]
- 49.Ban N., Nissen,P., Hansen,J., Capel,M., Moore,P.B. and Steitz,T.A. (1999) Placement of protein and RNA structures into a 5 Å-resolution map of the 50S ribosomal subunit. Nature, 400, 841–847. [DOI] [PubMed] [Google Scholar]