

Abstract
The title compound, C8H8N2S, was prepared by the condensation of N-methyl-1,2-phenylenediamine and carbon disulfide. The crystal structure is stabilized by a C—H⋯π interaction between a benzene H atom and the benzene ring of a neighbouring molecule, and by intermolecular N—H⋯S interactions.
Related literature
For related literature, see: Baily et al. (1996 ▶); Koch (2001 ▶); Namgun et al. (2001 ▶); Schuster et al. (1990 ▶); Patel & Chedekel (1984 ▶).
Experimental
Crystal data
C8H8N2S
M r = 164.22
Monoclinic,
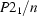
a = 9.997 (4) Å
b = 5.8140 (7) Å
c = 13.703 (4) Å
β = 94.05 (3)°
V = 794.5 (4) Å3
Z = 4
Mo Kα radiation
μ = 0.34 mm−1
T = 293 (2) K
0.20 × 0.10 × 0.02 mm
Data collection
Refinement
R[F 2 > 2σ(F 2)] = 0.029
wR(F 2) = 0.077
S = 1.09
962 reflections
101 parameters
H-atom parameters constrained
Δρmax = 0.20 e Å−3
Δρmin = −0.19 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2004 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2004 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2003 ▶); software used to prepare material for publication: PLATON.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808015043/lx2055sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808015043/lx2055Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2⋯Si | 0.86 | 2.57 | 3.408 (2) | 166 |
| C3—H3⋯Cgii | 0.93 | 2.74 | 3.464 (3) | 136 |
Symmetry codes: (i) 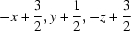 ; (ii)
; (ii) 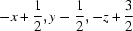 . Cg is the centroid of the C2–C7 ring.
. Cg is the centroid of the C2–C7 ring.
Acknowledgments
The authors acknowledge the Higher Education Commission, Pakistan, for financial support.
supplementary crystallographic information
Comment
N,N'- disubstituted and N-substituted thiourea derivatives are the major building blocks of organic macromolecular compounds. Thiourea derivatives such as benzothiazoles have been isolated by bromination of arylthioureas (Patil & Chedekel, 1984) and by condensation of 2-aminothiazole (Baily et al., 1996), by cyclization of N-(2-hydroxyethyl-N-methylthioureas and 2-methyl-aminothiazole (Namgun et al., 2001). Aliphatic and acylthioureas have a wide range of application due to their coordination behavior towards transition metals (Schuster et al., 1990). N,N-dialkyl-N-arylthioureas have been used for the extraction of metals such as nickel, palladium and platinum (Koch, 2001). Here we report the crystal structure of the title compound, 1-methyl-2H-benzimidazole-2-thione (Fig. 1).
The benzimidazole unit is essentially planar, with a mean deviation of 0.023 Å from the least-squares plane defined by the nine constituent atoms. The molecular packing (Fig. 2) is stabilized by a C—H···π interaction between a benzene H atom and the benzene ring of neighbouring molecules, with a C3—H3···Cgi separation of 2.735 (3) Å (Fig. 2 and Table 1; Cg is the C2-C7 benzene ring, symmetry code as in Fig. 2). Additionally, intermolecular N—H···S interactions in the structure were observed (Fig. 2 and Table 1; symmetry code as in Fig. 2).
Experimental
Compound (I) was synthesized by the addition of carbondisulfide (0.79 ml, 13.02 mmol) to N-methyl-1,2-phenylenediamine (0.744 ml, 6.55 mmol) in methanol (20 ml). The resulting mixture was stirred for 24 h, at 0°C temperature, giving a clear light yellow solution. The solution was evaporated under reduced pressure to give a light yellow solid, which was recrystallized in methanol/peteroleum ether (9:1) to afford compound (I) (yield : 76%).
Refinement
All H atoms were placed in idealized positions (C—H = 0.96 A ° (methyl); C—H = 0.93 A ° (aromatic); N—H = 0.86 A °) and refined as riding, with Uiso(H) = 1.2Ueq(C, N) or 1.5Ueq(C).
Figures
Fig. 1.

The structure of (I), with displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

C—H···π and N—H···S interactions (dotted lines) in the title compound. Cg denotes the ring centroid. [Symmetry code: (i) -x+1/2, y-1/2, -z+3/2; (ii) -x+3/2, y+1/2, -z+3/2; (iii) -x+3/2, y-1/2, -z+3/2; (iv) -x+1/2, y+1/2, -z+3/2.]
Crystal data
| C8H8N2S | F000 = 344 |
| Mr = 164.22 | Dx = 1.373 Mg m−3 |
| Monoclinic, P21/n | Melting point: 402 K |
| Hall symbol: -P_2yn | Mo Kα radiation λ = 0.71073 Å |
| a = 9.997 (4) Å | Cell parameters from 5701 reflections |
| b = 5.8140 (7) Å | θ = 3.7–23.1º |
| c = 13.703 (4) Å | µ = 0.34 mm−1 |
| β = 94.05 (3)º | T = 293 (2) K |
| V = 794.5 (4) Å3 | Block, colourless |
| Z = 4 | 0.20 × 0.10 × 0.02 mm |
Data collection
| Oxford Diffraction Xcalibur2 CCD diffractometer | 962 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 855 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.023 |
| Detector resolution: 10.0 pixels mm-1 | θmax = 23.1º |
| T = 293(2) K | θmin = 3.8º |
| ω scans | h = −10→11 |
| Absorption correction: analytical(CrysAlis RED; Oxford Diffraction; 2004; Clark & Reid, 1995) | k = −6→6 |
| Tmin = 0.929, Tmax = 0.967 | l = −14→14 |
| 7237 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.029 | H-atom parameters constrained |
| wR(F2) = 0.078 | w = 1/[σ2(Fo2) + (0.0353P)2 + 0.4983P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.09 | (Δ/σ)max = 0.001 |
| 962 reflections | Δρmax = 0.21 e Å−3 |
| 101 parameters | Δρmin = −0.19 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S | 0.73449 (6) | 0.62833 (10) | 0.62836 (4) | 0.0475 (3) | |
| N1 | 0.52959 (17) | 0.3736 (3) | 0.69273 (12) | 0.0345 (5) | |
| N2 | 0.57646 (17) | 0.6789 (3) | 0.78071 (12) | 0.0380 (5) | |
| H2 | 0.6119 | 0.8071 | 0.8004 | 0.046* | |
| C1 | 0.6119 (2) | 0.5599 (4) | 0.70145 (15) | 0.0359 (6) | |
| C2 | 0.4462 (2) | 0.3690 (3) | 0.77058 (15) | 0.0323 (5) | |
| C3 | 0.3528 (2) | 0.2095 (4) | 0.79703 (16) | 0.0399 (6) | |
| H3 | 0.3342 | 0.0773 | 0.7603 | 0.048* | |
| C4 | 0.2882 (2) | 0.2566 (4) | 0.88107 (17) | 0.0461 (6) | |
| H4 | 0.2255 | 0.1524 | 0.9017 | 0.055* | |
| C5 | 0.3145 (2) | 0.4548 (4) | 0.93499 (17) | 0.0466 (6) | |
| H5 | 0.2684 | 0.4812 | 0.9906 | 0.056* | |
| C6 | 0.4079 (2) | 0.6150 (4) | 0.90824 (16) | 0.0412 (6) | |
| H6 | 0.4246 | 0.7492 | 0.9440 | 0.049* | |
| C7 | 0.4752 (2) | 0.5656 (4) | 0.82572 (15) | 0.0330 (5) | |
| C8 | 0.5331 (3) | 0.1997 (4) | 0.61682 (17) | 0.0518 (7) | |
| H8A | 0.5508 | 0.2717 | 0.5560 | 0.078* | |
| H8B | 0.4482 | 0.1221 | 0.6098 | 0.078* | |
| H8C | 0.6026 | 0.0904 | 0.6345 | 0.078* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S | 0.0446 (4) | 0.0454 (4) | 0.0547 (4) | 0.0026 (3) | 0.0184 (3) | 0.0102 (3) |
| N1 | 0.0345 (10) | 0.0313 (10) | 0.0384 (10) | 0.0019 (8) | 0.0071 (8) | −0.0027 (8) |
| N2 | 0.0379 (11) | 0.0323 (10) | 0.0443 (11) | −0.0042 (8) | 0.0064 (9) | −0.0040 (9) |
| C1 | 0.0350 (12) | 0.0332 (12) | 0.0396 (13) | 0.0068 (11) | 0.0026 (10) | 0.0050 (10) |
| C2 | 0.0283 (11) | 0.0339 (13) | 0.0348 (12) | 0.0062 (10) | 0.0024 (9) | 0.0023 (10) |
| C3 | 0.0365 (12) | 0.0356 (13) | 0.0478 (14) | −0.0024 (11) | 0.0043 (11) | −0.0016 (11) |
| C4 | 0.0380 (13) | 0.0483 (16) | 0.0529 (15) | −0.0053 (11) | 0.0094 (12) | 0.0080 (13) |
| C5 | 0.0425 (13) | 0.0601 (16) | 0.0383 (13) | 0.0015 (13) | 0.0104 (11) | 0.0021 (12) |
| C6 | 0.0423 (13) | 0.0431 (14) | 0.0380 (13) | 0.0040 (11) | 0.0017 (10) | −0.0060 (11) |
| C7 | 0.0302 (12) | 0.0330 (12) | 0.0358 (12) | 0.0021 (10) | 0.0019 (10) | 0.0020 (10) |
| C8 | 0.0536 (15) | 0.0493 (15) | 0.0542 (15) | −0.0015 (13) | 0.0150 (12) | −0.0150 (13) |
Geometric parameters (Å, °)
| S—C1 | 1.684 (2) | C3—H3 | 0.9300 |
| N1—C1 | 1.361 (3) | C4—C5 | 1.384 (3) |
| N1—C2 | 1.400 (3) | C4—H4 | 0.9300 |
| N1—C8 | 1.453 (3) | C5—C6 | 1.386 (3) |
| N2—C1 | 1.356 (3) | C5—H5 | 0.9300 |
| N2—C7 | 1.389 (3) | C6—C7 | 1.386 (3) |
| N2—H2 | 0.8600 | C6—H6 | 0.9300 |
| C2—C3 | 1.383 (3) | C8—H8A | 0.9600 |
| C2—C7 | 1.389 (3) | C8—H8B | 0.9600 |
| C3—C4 | 1.387 (3) | C8—H8C | 0.9600 |
| C1—N1—C2 | 109.71 (17) | C3—C4—H4 | 119.2 |
| C1—N1—C8 | 124.88 (18) | C4—C5—C6 | 121.6 (2) |
| C2—N1—C8 | 125.34 (18) | C4—C5—H5 | 119.2 |
| C1—N2—C7 | 110.71 (18) | C6—C5—H5 | 119.2 |
| C1—N2—H2 | 124.6 | C7—C6—C5 | 116.8 (2) |
| C7—N2—H2 | 124.6 | C7—C6—H6 | 121.6 |
| N2—C1—N1 | 106.62 (18) | C5—C6—H6 | 121.6 |
| N2—C1—S | 126.72 (18) | C6—C7—N2 | 132.5 (2) |
| N1—C1—S | 126.65 (17) | C6—C7—C2 | 121.3 (2) |
| C3—C2—C7 | 121.86 (19) | N2—C7—C2 | 106.21 (17) |
| C3—C2—N1 | 131.46 (19) | N1—C8—H8A | 109.5 |
| C7—C2—N1 | 106.66 (17) | N1—C8—H8B | 109.5 |
| C2—C3—C4 | 116.6 (2) | H8A—C8—H8B | 109.5 |
| C2—C3—H3 | 121.7 | N1—C8—H8C | 109.5 |
| C4—C3—H3 | 121.7 | H8A—C8—H8C | 109.5 |
| C5—C4—C3 | 121.7 (2) | H8B—C8—H8C | 109.5 |
| C5—C4—H4 | 119.2 | ||
| C7—N2—C1—N1 | 2.4 (2) | C2—C3—C4—C5 | 0.9 (3) |
| C7—N2—C1—S | −177.28 (16) | C3—C4—C5—C6 | −0.7 (4) |
| C2—N1—C1—N2 | −3.2 (2) | C4—C5—C6—C7 | −1.0 (3) |
| C8—N1—C1—N2 | 179.79 (19) | C5—C6—C7—N2 | −177.1 (2) |
| C2—N1—C1—S | 176.50 (15) | C5—C6—C7—C2 | 2.4 (3) |
| C8—N1—C1—S | −0.5 (3) | C1—N2—C7—C6 | 178.9 (2) |
| C1—N1—C2—C3 | −175.6 (2) | C1—N2—C7—C2 | −0.7 (2) |
| C8—N1—C2—C3 | 1.4 (3) | C3—C2—C7—C6 | −2.3 (3) |
| C1—N1—C2—C7 | 2.8 (2) | N1—C2—C7—C6 | 179.11 (19) |
| C8—N1—C2—C7 | 179.79 (19) | C3—C2—C7—N2 | 177.32 (19) |
| C7—C2—C3—C4 | 0.6 (3) | N1—C2—C7—N2 | −1.2 (2) |
| N1—C2—C3—C4 | 178.8 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Si | 0.86 | 2.57 | 3.408 (2) | 166 |
| C3—H3···Cgii | 0.93 | 2.74 | 3.464 (3) | 136 |
Symmetry codes: (i) −x+3/2, y+1/2, −z+3/2; (ii) −x+1/2, y−1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: LX2055).
References
- Baily, N., Dean, A. W., Judd, D. B., Middlemiss, D., Storer, R. & Watson, S. P. (1996). Bioorg. Med. Chem. Lett.6, 1409–1413.
- Clark, R. C. & Reid, J. S. (1995). Acta Cryst. A51, 887–897.
- Koch, K. R. (2001). Coord. Chem. Rev.216, 473–482.
- Namgun, L., Mi-Hyun, C. & Tack, H. K. (2001). J. Korean Chem. Soc.45, 96–99.
- Oxford Diffraction (2004). CrysAlis CCD and CrysAlis RED. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England.
- Patel, D. G. & Chedekel, M. R. (1984). J. Org. Chem.49, 997–1000.
- Schuster, M., Kugler, B. & Konig, K. H. (1990). J. Anal. Chem.338, 717–720.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808015043/lx2055sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808015043/lx2055Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report