


Abstract
Tumor necrosis factor α (TNFα) acts as a beneficial mediator in the process of host defence. In recent years major interest has focused on the AU-rich elements (AREs) present in the 3′-untranslated region (3′-UTR) of TNFα mRNA as this region plays a pivotal role in post-transcriptional control of TNFα production. Certain stimuli, such as lipopolysaccharides, a component of the Gram-negative bacterial cell wall, have the ability to relinquish the translational suppression of TNFα mRNA imposed by these AREs in macrophages, thereby enabling the efficient production of the TNFα. In this study we show that the polymorphism (GAU trinucleotide insertional mutation) present in the regulatory 3′-UTR of TNFα mRNA of NZW mice results in the hindered binding of RNA-binding proteins, thereby leading to a significantly reduced production of TNFα protein. We also show that the binding of macrophage proteins to the main ARE is also decreased by another trinucleotide (CAU) insertion in the TNFα 3′-UTR. One of the proteins affected by the GAU trinucleotide insertional mutation was identified as HuR, a nucleo-cytoplasmic shuttling protein previously shown to play a prominent role in the stability and translatability of mRNA containing AREs. Since binding of this protein most likely modulates the stability, translational efficiency and transport of TNFα mRNA, these results suggest that mutations in the ARE of TNFα mRNA decrease the production of TNFα protein in macrophages by hindering the binding of HuR to the ARE.
INTRODUCTION
Tumor necrosis factor α (TNFα) is a key pro-inflammatory cytokine, produced predominately by monocytes and macrophages, which exerts pleiotropic effects on numerous cell types (1–3). TNFα is an important mediator of both specific and non-specific immune responses. Excessive production of TNFα due to global infections (which occur in burned or immunocompromized patients) or other conditions such as trauma or severe tissue injury results in systemic inflammatory response syndrome (SIRS), which is characterized by fever, increased vasodilatation, intravascular coagulation and haemorrhagic necrosis (resulting in a circulatory collapse and multiple organ failure) (4–7).
Although the production of TNFα is controlled transcriptionally by various stimuli, such as lipopolysaccharide (LPS; a component of the Gram-negative bacterial cell wall) and interferon γ (IFNγ), a number of studies have recently demonstrated that production of TNFα is also regulated post-transcriptonally (8–10). Regulation at the post-transcriptional level involves the regulation of both the mRNA stability and translational efficacy (10–14). The sequences in the 3′-untranslated region (3′-UTR) of TNFα mRNA responsible for the translational repression and inducibility by LPS are located between bases 1195 and 1374 of the TNFα mRNA 3′-UTR. This region is characterized by multiple repeats of an AUUUA motif (AU-rich element, ARE) (9). AREs are also found in several early response genes (cytokines and protooncogenes) whose mRNA is very unstable (15–18). The AREs of several genes are known to be the recognition sequences for several RNA-binding proteins (19–21). RNase protection and RNA gel shift assays have previously enabled us to map two protein binding regions of TNFα mRNA necessary for binding of macrophage proteins to the 3′-UTR (22,23). The first protein binding site present within this main ARE is positioned between bases 1291 and 1329, whereas the second protein binding site (located 147 bases downstream from the first ARE) is 31 nt long and contains a single AUAUUUAU sequence.
NZW mice have previously been reported to be low producers of TNFα protein when peritoneal macrophages from these mice are stimulated with IFNγ and LPS (24–26). F1 mice, obtained when NZW mice are bred with NZB mice, (due to a decreased production of TNFα protein by NZW mice) have been shown to develop pathologies similar to what is seen in humans who develop systemic lupus erythematosus (SLE) (27,28). The onset of pathologies (i.e. nephritis) associated with this autoimmune disease is delayed by injection of the mice with recombinant TNFα or with AS101 (a synthetic immunomodulator capable of inducing the production of TNFα) (27,29). NZW mice have been shown to contain a mutation insertion in the main ARE of TNFα mRNA 3′-UTR (30,31). Jacob et al. (30) have demonstrated with the use of reporter constructs that this mutation affects the post-transcriptional regulation of TNFα production.
Recently, proteins capable of binding to the main ARE of TNFα mRNA 3′-UTR have been identified in both macrophage and non-macrophage cells. Gueydan et al. (32) have reported that TIAR, a known RNA-binding protein (33), is able to bind to the TNFα mRNA 3′-UTR ARE. This protein is localized to the cytoplasm of murine macrophages and is present in a complex that is not inducible by LPS. Tristetrapolin (a CCCH zinc finger protein present in macrophages) has also been shown to interact with the ARE present in the 3′-UTR of TNFα mRNA. This protein is inducible by LPS in macrophages and has been shown to be involved in the destabilization of TNFα message (34,35). Lastly, ELAV-like proteins, detected in both human hippocampal tissue and neuroblastoma cell line extract, are also able to bind to the main ARE in the 3′-UTR of TNFα message (36). HuR, a member of the ELAV-like family of proteins, is ubiquitously expressed in all cells (37). This protein, which contains three RNA recognition motifs, has been shown to possess a nuclear–cytoplasmic shuttling sequence that may be essential in the transport and protection of RNA containing ARE (37,38).
The primary goal of this study was to investigate whether the trinucleotide insertion in the 3′-UTR of NZW mice hinders the binding of macrophage proteins to the ARE region. We therefore assessed if this mutation, which affects the post-transcriptional regulation of TNFα, has any consequences on the binding of macrophage proteins to the main ARE element (which has been shown to play a predominant role in the stability and translatability of TNFα mRNA). We show that trinucleotide insertions in the main ARE of the 3′-UTR of TNFα mRNA alter the binding of complexes B and C to the region. These complexes, present in the nucleus and associated with polyribosomes in the cytoplasm, may be involved in the transport of TNFα mRNA from the nucleus to the cytoplasm. We have identified one of the proteins in the affected complexes as being the ELAV-like protein HuR, a nucleo-cytoplasmic shuttling protein that has previously been shown to regulate the stability of mRNA containing ARE. This is the first report that shows that HuR binds to the ARE in the 3′-UTR of TNFα mRNA in macrophages. The decreased production of TNFα protein by peritoneal macrophages obtained from NZW mice, therefore, may be due to the decreased binding of a complex containing HuR to the mutated message, which consequently results in the hindered transport of TNFα mRNA from the nucleus to the cytoplasm.
MATERIALS AND METHODS
Reagents
The following reagents were used: heparin sulphate, proteinase K, Nonidet P-40 (NP-40) and polymethylsulfonylfluoride from Sigma (St Louis, MO), [α-32P]UTP and [α-32P]ATP from Amersham (Amersham Canada, Oakville, Ontario), DNase I, aprotinin, leupeptin, RNase inhibitor and SP6 RNA polymerase from Pharmacia (Piscataway, NJ), RNase T1 and T7 RNA polymerase from USB/Amersham (Cleveland, OH).
Mice
Female or male NZW.Lac J mice were purchased from Jackson Laboratories. The B10.A mice (carrying the wild-type allele of the Nramp 1 gene) were bred in the Montreal General Hospital Research Institute animal facility under specific-pathogen-free conditions. These mice were injected intraperitoneally with 1 ml of 4% thioglycollate medium and peritoneal exudate cells were harvested 4 days later by washing three times with Hanks’ balanced salt solution (HBSS; Life Technologies, Grand Island, NY). The peritoneal exudate cells were washed three times with HBSS and the macrophages were purified by adherence to plastic. For analyses of TNFα protein, cells maintained in RPMI medium 1640 (Life Technologies, Grand Island, NY) supplemented with 5% fetal bovine serum (HyClone, Logan, UT) were plated at a concentration of 2 million/ml, washed with HBSS after adherence of the cells and then stimulated with IFNγ (100 U/ml) and/or LPS (1 µg/ml) for 16 h.
Cells
Murine macrophage cell line B10R, derived from the bone marrow of B10A mice (wt Nramp 1) was described in detail previously (39). The cells were maintained in Dulbecco’s modified minimal essential medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml of streptomycin.
TNFα ELISA
The concentration of TNFα was determined by sandwich ELISA as described by Sheehan et al. (40). Briefly, ELISA microplates were coated with hamster anti-murine TNFα mAb (Genzyme), and sequentially incubated with various dilutions of sample, polyclonal rabbit anti-murine TNFα anti-serum, peroxidase-conjugated goat anti-rabbit IgG antibodies (Bio-Rad, Hercules, CA) and peroxidase substrate (ECL, Buckinghamshire, UK). The intensity of the colorimetric reaction was determined by spectrometry at 405 nm. TNFα concentration was calculated with reference to a standard curve established with recombinant TNFα (Genzyme).
Preparation of RNA transcripts
Construct TNF-9 was obtained by subcloning the RT–PCR-amplified 3′-UTR of murine (strain C57Bl/6) TNFα (bases 823–1611, DDBJ/GenBank/EMBL accession no. M11731) into vector pGEM3Zf(+) (Promega). The TNF-9m probe was obtained by subcloning the 3′-UTR obtained from NZW mice into the T-easy vector (Promega). TNF-10 (containing the first main ARE) as well as the TNF-10m and TNF-10m2 probes were transcribed from synthesized oligonucleotides which were subcloned into pGEM3Zf(+) (Promega). Clones were linearized 3′ of the ARE prior to their use in the transcription reaction. Transcription reactions were performed according to a modification of the protocol by Gurevich et al. (41). The transcription with T7 RNA polymerase was performed in the reaction mix containing 80 mM HEPES pH 7.5, 2 mM MgCl2, 2 mM spermidine, 30 mM dithiothreitol (DTT), 0.5 mM of each ATP, GTP and CTP, 12 µM UTP, 40 µCi [α-32P]UTP (3000 Ci/mmol), 50 µg/ml of plasmid DNA and 500 U/ml of T7 polymerase for 30 min at 37°C. The size and integrity of cRNA transcripts was verified by electrophoresis on polyacrylamide gel containing 6 M urea.
Preparation of cellular extracts
The procedure for the preparation of cellular extracts obtained from either untreated or Actinomycin D treated cells (1 h treatment; 5 µg/ml) is based on that described previously (21,22,42–45). The cells were cooled on ice for 5 min, centrifuged at 150 g for 5 min and resuspended in ice-cold EBKL buffer containing 25 mM HEPES pH 7.6, 5 mM MgCl2, 1.5 mM KCl, 2 mM DTT, protease inhibitors (1 mM PMSF, 4 µg/ml of each aprotinin and leupeptin) and 0.1% NP-40. The cells were then lysed on ice by 20 strokes in a Dounce homogenizer (Sigma; tight pestle). The nuclei were removed by two 3 min centrifugations at 600 g. The resulting supernatant, spun at 10 000 g for 10 min, was labeled as the total cytoplasmic extract.
The nuclei obtained in the initial step of fractionation were washed first in EBMK buffer, spun at 150 g, and then washed with EBMK buffer containing 0.5% NP-40. The supernatant from this step was designated the outer nuclear membrane wash fraction. The nuclei were incubated in EBKL (0.1% NP-40) for 10 min and lysed by dropwise addition of KCl to 0.2 M final concentration. The lysed nuclei were spun at 10 000 g for 10 min. The supernatant obtained was designated the nucleoplasm. Total cellular extract was prepared by the sonication of lysed cells (3 × 10 s at 200 W) in EBKL buffer containing 0.1% NP-40.
The RNA-binding assay
The protein extracts were incubated with the indicated cRNA probes in buffer containing 20 mM HEPES pH 7.6, 75 mM NaCl, 1.5 mM KCl, 5 mM MgCl2, 175 mM sucrose, 2 mM DTT and protease inhibitors (1 mM PMSF, 4 µg/ml of each aprotinin and leupeptin). Typically, 10 µg of protein were incubated with 200 000 c.p.m. of 32P-labeled cRNA TNF-9 probe in a total volume of 20 µl. The reaction was carried out at room temperature for 15 min. RNase T1 and heparin sulphate were added to final concentrations of 3000 U/ml and 5 mg/ml, respectively, and the mixture was incubated for an additional 15 min. The cRNA was then covalently crosslinked to the proteins by an 8 min exposure to shortwave UV light (254 nm) at 4 cm distance using Stratalinker (Stratagene, La Jolla, CA; energy 250 J/cm2). The samples were resolved either on a 4% polyacrylamide gel containing 0.05% NP-40 or by 8% SDS–PAGE. 32P-labeled TNF-10/m/m2 probes (100 000 c.p.m.) were used in binding reactions. In the supershift experiments, 5 µg of a mAb (19F12) directed against HuR was added to the samples 15 min after the addition of RNase T1. The reaction was then incubated for an additional 20 min at room temperature prior to loading on the gel without UV crosslinking.
RESULTS
NZW mice are low producers of TNFα
As previously reported, NZW mice have been shown to have a trinucleotide (GAU) mutational insertion in the main ARE of the TNFα mRNA 3′-UTR and that this mutation results in decreased TNFα production (24,30,31). We sought to verify whether B10.A mice, which do not have a mutation in the 3′-UTR, produce more TNFα protein than NZW mice. We therefore performed ELISA analysis with supernatants obtained from thioglycollate-elicited peritoneal macrophages from both B10.A and NZW mice that were stimulated with LPS (1 µg/ml) and/or IFNγ (100 U/ml) for 16 h (Fig. 1). Supernatants from untreated cells were used as controls. We showed that B10.A peritoneal macrophages treated with LPS or IFNγ produced more TNFα protein (52.3 ± 8.4 and 25.2 ± 11.2 U/ml, respectively) than NZW mice (LPS, 21.4 ± 8.7 U/ml; IFNγ, 0.44 ± 0.3 U/ml). The most predominate difference was seen when the cells were treated with both LPS and IFNγ for 16 h. B10.A mice produced significantly more TNFα (94.1 ± 15.1 U/ml) than NZW mice (36.8 ± 9.5 U/ml) when peritoneal macrophages isolated from both mice strains were stimulated with both LPS and IFNγ for 16 h. B10.A mice (which do not have an insertional mutation in the TNFα mRNA 3′-UTR) are thus more efficient producers of TNFα protein than NZW mice.
Figure 1.
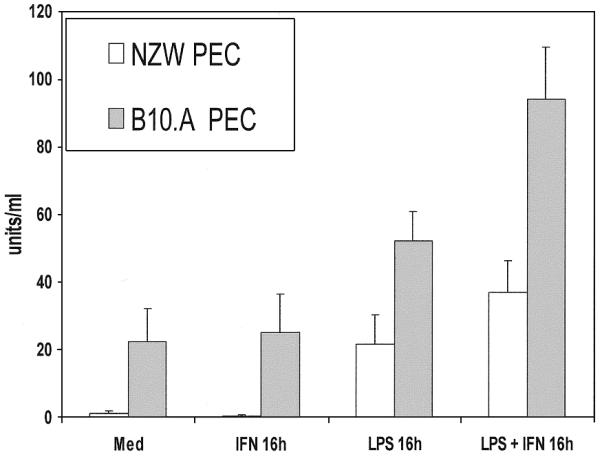
TNFα protein production is differentially regulated in peritoneal macrophages (activated with IFNγ and/or LPS) isolated from both B10.A and NZW mice. ELISA analysis was performed with supernatants obtained from peritoneal macrophages stimulated with LPS and/or IFNγ for 16 h. Supernatants obtained from untreated cells were used as a control. The concentrations of the stimuli were as follows: LPS, 1 µg/ml; IFNγ, 100 U/ml.
A trinucleotide mutational insertion hinders binding of macrophage proteins to NZW TNFα mRNA 3′-UTR
We have previously mapped two protein-binding regions of TNFα mRNA necessary for the binding of macrophage proteins to the 3′-UTR of the message (22,23). The first protein-binding region is located inside the main ARE of the TNFα 3′-UTR, while the second protein binding region is located 147 bases downstream of the first region. In NZW mice, a trinucleotide GAU insertion is found at the 5′ end of the first main ARE in the 3′-UTR of TNFα mRNA (Fig. 2). We therefore wanted to investigate the consequences of this polymorphism on the binding of regulatory proteins to this region. We tested whether there is a difference in the binding of macrophage proteins to the main ARE in the 3′-UTR of TNFα mRNA obtained from NZW mice in comparison to a non-mutated ARE in the 3′-UTR of TNFα mRNA found in B10.A mice. In order to determine whether there is a difference in RNA-binding activity, we performed RNA-binding assays with total cellular fractions obtained from untreated macrophages (Fig. 3). We observed that the macrophage complexes A, B and C bind to the full-length 3′-UTR cRNA probe TNF-9 containing a non-mutated ARE (lane 2) to a greater extent than to the TNF-9m cRNA probe containing mutated ARE from NZW TNFα 3′-UTR (lane 4). The binding of macrophage proteins to the 3′-UTR of NZW mRNA was hindered by a GAU insertional mutation in the first main ARE.
Figure 2.

Sequence and position of cRNA probes used in the study. (A) Position of cRNA probes of murine B10.A (TNF-9) and NZW (TNF-9m) TNFα 3′-UTR as well as the position of the synthetic TNF-10, TNF-10m, TNF-10m2 cRNA probes. ARE are shown as black boxes; the GAU insertional mutation in NZW ARE at position 1298 is also indicated. (B) Sequence of the synthetic probes encoding the first (TNF-10) protein binding region as well as the synthetic probes containing the trinucleotide insertions (TNF-10m, TNF-10m2).
Figure 3.
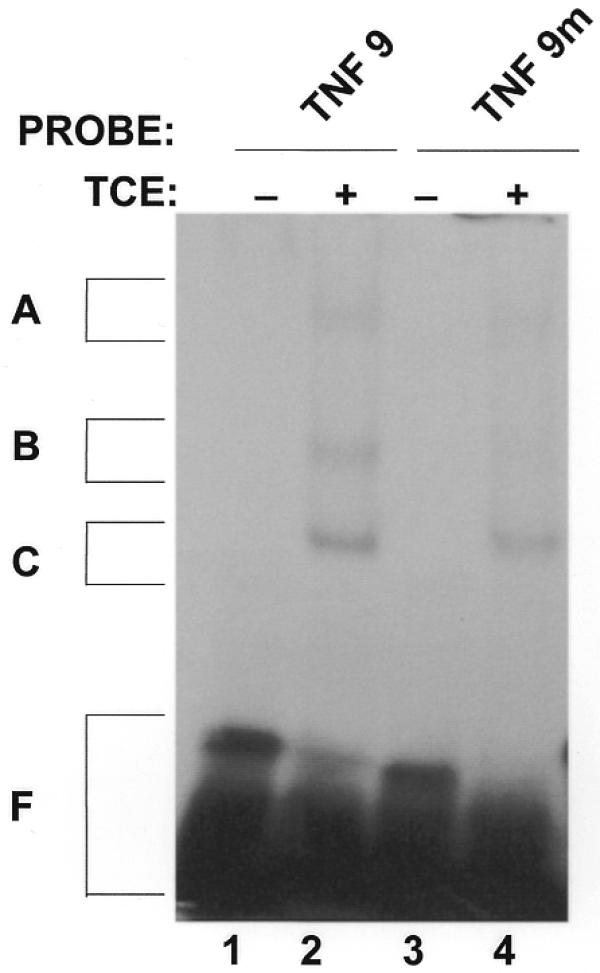
Binding of macrophage proteins to the 3′-UTR of TNFα mRNA isolated from NZW mice. Radiolabeled cRNA TNF-9 probe (200 000 c.p.m.; encompassing the whole TNFα mRNA 3′-UTR found in B10.A mice) as well as TNF-9m probe (containing 3′-UTR isolated from NZW mice with the GAU insertion in the main ARE) were incubated alone (lanes 1 and 3) or with 10 µg of total cellular extract (TCE) obtained from an untreated B10R cell line (lanes 2 and 4). The reaction mixture was incubated for 15 min at room temperature prior to treatment with RNase T1. The RNA–protein complexes were then UV-crosslinked for 8 min and the resulting complexes were analyzed by non-denaturing gel electrophoresis. A, B and C denote the positions of the main RNA–protein complexes. F denotes free nucleotides and remnants of undigested RNA.
Both GAU and CAU trinucleotide insertions hinder binding of macrophage proteins to the main ARE of TNFα 3′-UTR mRNA
In order to confirm that the decrease in binding of macrophage proteins to the 3′-UTR of NZW TNFα mRNA is due to a trinucleotide insertion in the main ARE, a double-stranded oligonucleotide encoding the first main 39 nt protein-binding region (designated TNF-10; Fig. 2) as well as an oligonucleotide identical to TNF-10 with a GAT (designated TNF-10m) or a CAT (designated TNF-10m2) insertion were synthesized. RNA-binding assays were then performed with cRNA probes derived from the three synthetic oligonucleotides (TNF-10/m/m2) (Figs 4A and 5A). As indicated in Materials and Methods, the ribonuclease RNase T1 is employed in the RNA-binding assay. The role of RNase T1 in this assay is to cleave any mRNA not protected by macrophage proteins, which in our case is outside the main ARE protein-binding region. RNase T1, however, cleaves predominately 3′ to guanine residues present in the RNA. RNA-binding assays were therefore performed without RNase T1 when experiments were done with the TNF-10m cRNA probe (with a GAU trinucleotide insertion in the main ARE). In these experiments, RNase T1 was also omitted from samples containing the TNF-10 probe in order to adequately compare intensities of complexes bound to the two probes TNF-10 and the TNF-10m. As shown in Figure 4A and summarized in Table 1, macrophage protein complexes B (66.4 ± 4.8% binding relative to TNF-10) and C (55.5 ± 4.6% binding relative to TNF-10) were hindered in the binding to the TNF-10m probe as a result of the GAU insertion. Using SDS–PAGE analysis we demonstrated that at least four RNA-binding proteins (of apparent molecular weights 48, 81, 89 and 125 kDa) bind less to the TNF-10m cRNA probe (Fig. 4B, lane 4) than to the TNF-10 probe (lane 2). Therefore, the GAU trinucleotide insertion hinders the binding of macrophage proteins to the ARE of TNFα mRNA. Similarly to the TNF-10m cRNA probe, we were also able to demonstrate a decrease in the binding of macrophage complexes B and C to the TNF-10m2 cRNA probe (which contains a CAU insertion) (Fig. 5A, lane 4). This was demonstrated by the fact that the intensities of the TNF-10m2 RNA–protein complexes relative to the TNF-10 probe were 88.9 ± 13.8, 67.2 ± 8.3 and 61.1 ± 3.6% for complexes A, B and C, respectively (Table 1). SDS–PAGE analysis showed that the interaction of at least three to four RNA-binding proteins was less abundant for the TNF-10m2 cRNA probe (Fig. 5B, lane 4) than for the TNF-10 probe (Fig. 5B, lane 2). Interestingly, the 48 kDa protein complex is most affected by trinucleotide insertions (GAU or CAU) in the main ARE of TNFα mRNA 3′-UTR (Figs 4B and 5B). Both GAU and CAU trinucleotide insertions prevented binding of macrophage proteins to the main ARE of TNFα 3′-UTR mRNA.
Figure 4.
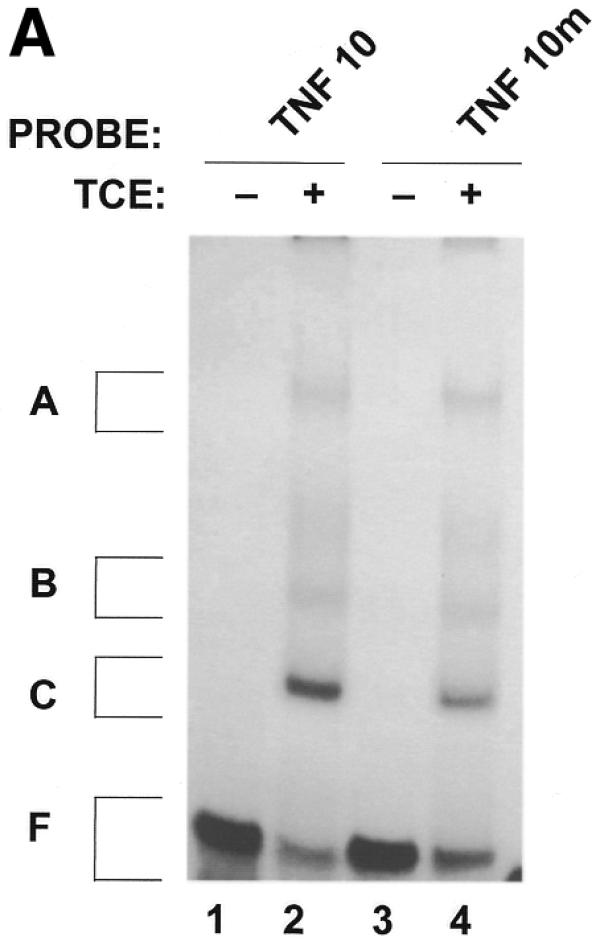
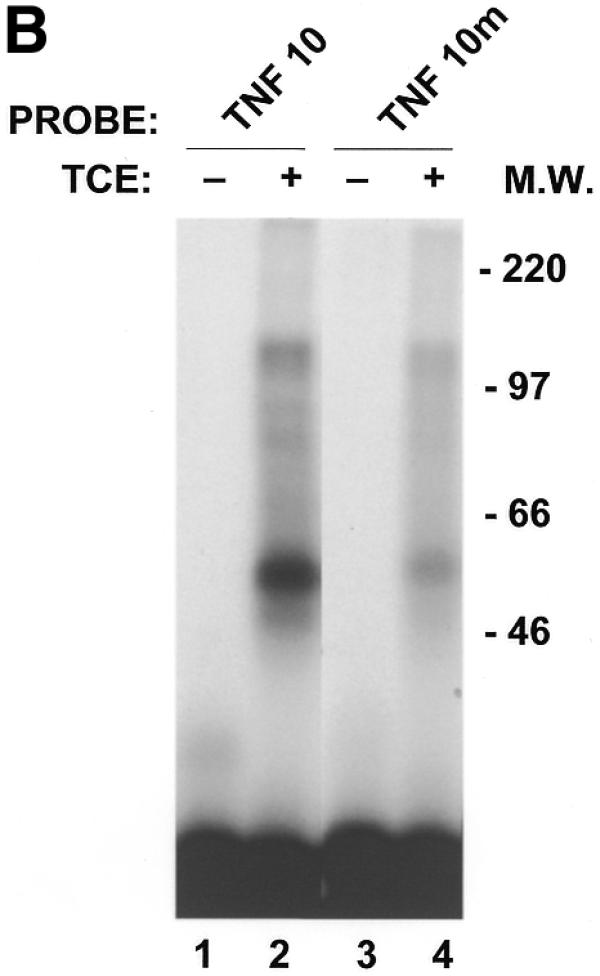
Binding of macrophage proteins to the TNFα ARE is hindered by a GAU trinucleotide insertion. RNA-binding assays were performed with 100 000 c.p.m. of labeled TNF-10 and TNF-10m (containing GAU insertion) cRNA probes, alone (lanes 1 and 3) or with 10 µg of TCE obtained from macrophage cells (lanes 2 and 4) in the absence of RNase T1. The RNA–protein complexes were then UV-crosslinked for 8 min and the resulting complexes were analyzed by non-denaturing gel (A) or by SDS–PAGE gels (B). A, B and C denote the positions of the main RNA–protein complexes. F denotes free probe and free nucleotides.
Figure 5.
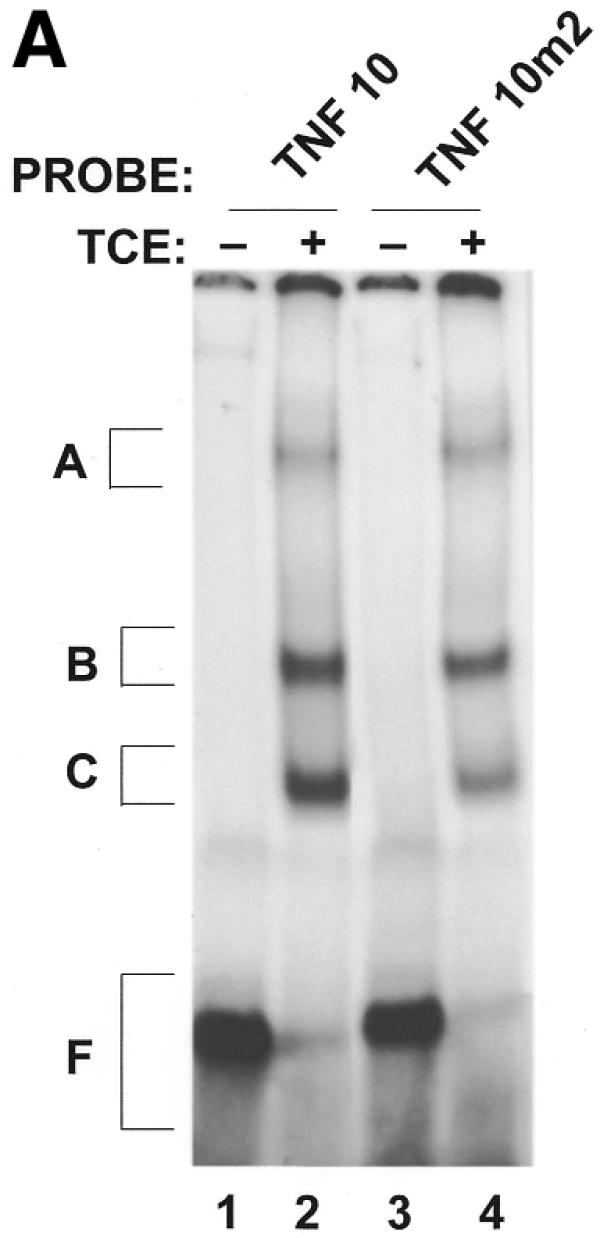
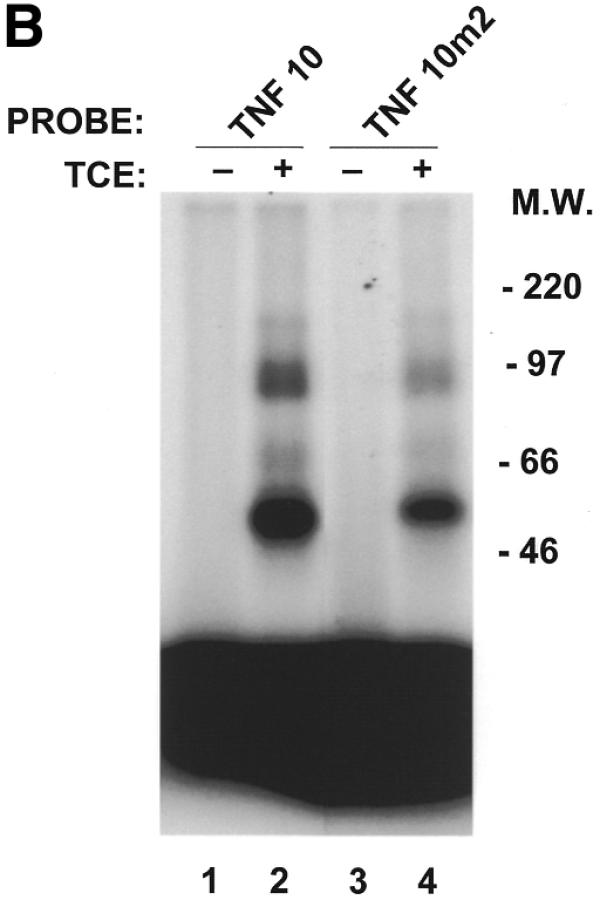
Binding of macrophage proteins to the TNFα ARE is hindered by a CAU trinucleotide insertion. cRNA labeled TNF-10 and TNF-10m2 (CAU insertion) probes (100 000 c.p.m.) were incubated alone (lanes 1 and 3) or with 10 µg of TCE prepared from untreated murine macrophages (lanes 2 and 4). The resulting RNA–protein complexes were treated with RNase T1, UV-crosslinked and analyzed on non-denaturing polyacrylamide gel (A) as well as on SDS–PAGE gels (B). A, B and C denote the positions of the main RNA–protein complexes. F denotes free probe, free nucleotides and remnants of undigested RNA.
Table 1. Intensities of the RNA–mutant probe complexes expressed as percentages relative to TNF-10–protein complexes.
| Mutant probe |
Complex A |
Complex B |
Complex C |
| TNF-10m | 82.1 ± 5.4 | 66.4 ± 4.8 | 55.5 ± 4.6 |
| TNF-10m2 | 88.9 ± 13.8 | 67.2 ± 8.3 | 61.1 ± 3.6 |
Experiments were performed with equal amounts of each of the cRNA probes. In order to further ensure that equal amount of probes were used in the reactions, data were normalized by determining the quantity of cRNA free probe present in the control lanes (without protein extract) of both the non-mutant (TNF-10) and the mutant (TNF-10m, TNF-10m2) probes. Quantification of the intensities of the complexes were then adjusted by taking into account differences in the free probes.
Results are representative of four independent experiments.
Actinomycin D induces nuclear and cytoplasmic RNA–protein complexes
Several studies have demonstrated that the stability of mRNAs containing an AU-rich region in their 3′-UTR was sensitive to inhibition of translation and transcription (46,47). Therefore, we addressed the question of whether transcriptional inhibitors would modulate the levels or localization of TNFα mRNA-binding proteins. As shown in Figure 6A and B, treatment of cells with the transcriptional inhibitor Actinomycin D enhanced the formation of protein complexes B and C in the nuclear wash (Fig. 6A, lanes 4 and 5) as well as in both the nucleoplasmic (Fig. 6A, lanes 6 and 7) and cytoplasmic extracts (Fig. 6A, lanes 8 and 9). SDS–PAGE analyses indicated that Actinomycin D dramatically increased the formation of a 48 kDa RNA–protein in the nuclear and cytoplasmic extracts of treated cells (Fig. 6B). Actinomycin D, therefore, affects the cellular localization of complexes whose binding activities are hindered by mutations in the ARE of the TNFα 3′-UTR.
Figure 6.
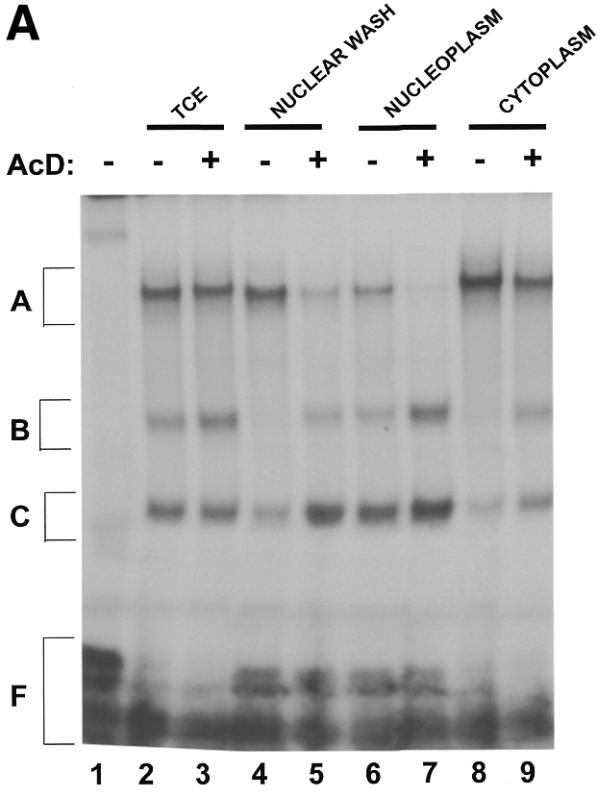
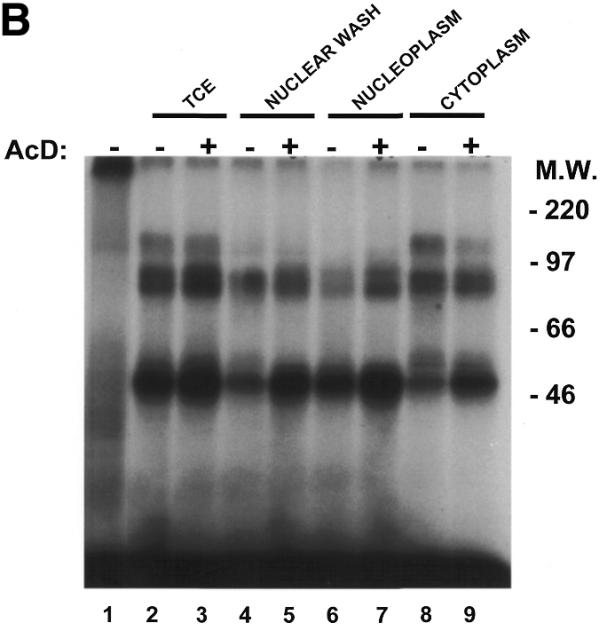
Actinomycin D induces the formation of complex B and C with TNFα mRNA. Probe TNF-10 was incubated alone (lane 1) or with protein extracts prepared from B10R cells untreated (lanes 2, 4, 6 and 8) or treated with 5 µg/ml of Actinomycin D for 1 h (lanes 3, 5, 7 and 9). Lanes 2 and 3, total cellular extract; lanes 4 and 5, nuclear wash fraction; lanes 6 and 7, nucleoplasm; lanes 8 and 9, total cytoplasmic extract. The resulting RNA–protein complexes were analyzed either on the non-denaturing gel (A) or UV-crosslinked and analyzed on denaturing 8% polyacrylamide–SDS gel (B).
HuR is present in complexes B and C
The ELAV-like mRNA-binding proteins have previously been shown to bind the ARE of TNFα mRNA (36). These proteins have also been shown to be associated with polyribosomes and are believed to be involved in mRNA transport (48). One of these proteins, HuR, whose localization in the cell has been shown to be affected by Actinomycin D (49), is known to increase the stability of messages containing ARE (49,50). Since HuR plays a prominent role in the post-transcriptional regulation of mRNA containing AREs, we wanted to verify whether HuR is present in any of the complexes affected by the insertional mutation in the ARE of the TNFα message. We therefore performed RNA–protein supershift experiments with a monoclonal antibody directed against HuR (Fig. 7). We observed that the mAb (when added to the samples containing TNF-10 cRNA probe and total cellular extract) was able to shift both complexes B and C (Fig. 7, lane 3). We also demonstrated that the levels of the shifted complex in total cytoplasmic extract was increased when cells were treated with Actinomycin D (Fig. 7, lane 7) compared to untreated cells (Fig. 7, lane 5). These results therefore indicate that HuR, present in complex C, localizes to the cytoplasm when cells are treated with Actinomycin D. Interestingly, we also observed that binding of the shifted complex (therefore containing HuR) to the TNF-10m cRNA was less than to the TNF-10 cRNA probes (data not shown). Consistent with data illustrated in Figure 4A, scanning results indicated that the mutation caused a 35% decrease in the binding of complexes containing HuR.
Figure 7.
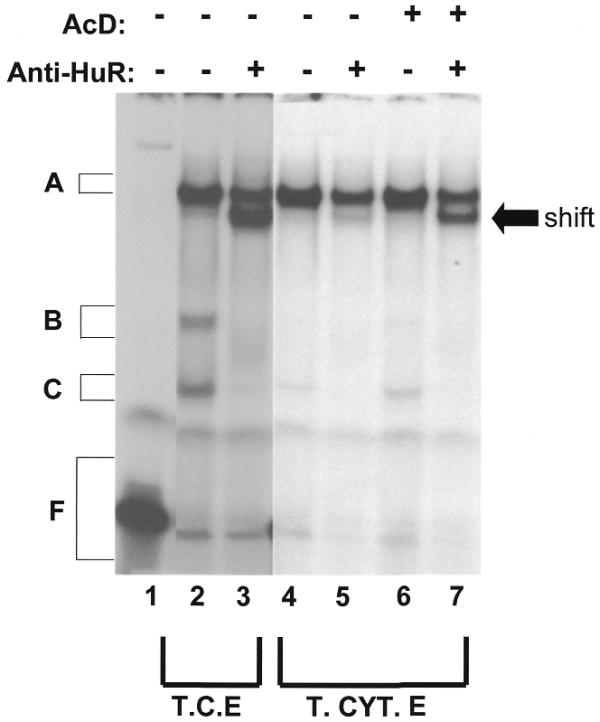
HuR protein is present in complexes B and C. TNF-10 cRNA probe (100 000 c.p.m.) was incubated alone (lane 1) or with 10 µg of total cellular extract (lanes 2 and 3) or with 10 µg of total cytoplasmic extract (lanes 4–7) for 15 min. The cytoplasmic extracts used in the assay were obtained from cells treated with (lanes 6 and 7) or without (lanes 4 and 5) Actinomycin D (AcD; 5µg/ml) for 1 h. Following treatment of the complexes with RNase T1, the 19F12 mAb was added to samples (lanes 3, 5 and 7) for 20 min at room temperature. The samples with no UV-crosslinking where then loaded on a 4% non-denaturing polyacrylamide gel.
DISCUSSION
The aim of this study was to investigate whether a GAU insertional mutation in the main ARE of the 3′-UTR of TNFα mRNA has an effect on the binding of macrophage proteins to this element. We also wanted to identify proteins whose binding activity is affected by this mutation in order to better understand the mechanisms underlying the post-transcriptional control of TNFα gene expression. Since this polymorphism was found in NZW mice, we wanted to verify whether mice that do not contain this mutation, such as B10.A mice, produce more TNFα than mice that contain this mutation. Our results show that peritoneal macrophages isolated from B10.A mice and stimulated with LPS and/or IFNγ for 16 h produce more TNFα protein than NZW mice (Fig. 1). The production is significantly greater when the macrophages are stimulated with IFNγ alone, LPS alone or with both stimuli together. Although there is a difference between the two strains, both NZW and B10.A mice synergistically upregulate TNFα production when the cells are treated with both IFNγ and LPS for 16 h in comparison to both stimuli alone. Jacob and Tashman (24) previously showed that strains that are classified as low producers of TNFα protein contain a GAU trinucleotide insertion in the ARE of TNFα mRNA 3′-UTR. Schook et al. (51) did not find a significant difference in the LPS-induced TNFα production in bone marrow-derived or thioglycollate-elicited macrophages from the NZW and C57Bl/6 strains of mice. However, the production of TNFα following treatment with IFNγ and LPS was not assessed in their studies. Since we show that the biggest difference can be observed when the macrophages are treated with IFNγ and LPS, the results presented in our manuscript and published previously by Jacob and Tashman (24) cannot be adequately compared to the results published by Schook et al. (51). Using luciferase reporter constructs, Jacob and colleagues also demonstrated that there is no significant difference in transcriptional activity between high and low TNFα producing mouse strains (30). They furthermore proved through the use of reporter constructs containing the TNFα mRNA 3′-UTR derived from high or low TNFα producing mouse strains that the decrease in TNFα levels in NZW mice is indeed due to the trinucleotide insertion the main ARE of the message (30). Overall, these authors demonstrated that the mutation post-transcriptionally regulated TNFα. Bazzoni and Beutler (52) have demonstrated, by RT–PCR, that F1(NZW×NZB) mice produce equal quantities of TNFα mRNA encoded from both NZW and NZB alleles. These data also suggested that the low production of TNFα protein in the F1(NZB×NZW) mice occurs at the post-transcriptional level, most likely affecting TNFα mRNA association with polyribosomes and/or translational repression. The precise molecular mechanism by which this polymorphism in NZW mice affects the post-transcriptional regulation of TNFα remains to be established.
We have previously mapped two regions within the 3′-UTR of TNFα mRNA capable of binding to macrophage proteins (22,23). The existence of the two protein-binding regions in the TNFα 3′-UTR support data by Han and colleagues (9) who provide evidence (by sequentially deleting various parts of the TNFα 3′-UTR linked to a CAT reporter gene) that other sequences in the 3′-UTR may act synergistically with the first main ARE in the control of the translational repression and inducibility of TNFα. We therefore decided to verify whether the binding of RNA-binding proteins to the main ARE was affected by the trinucleotide insertional mutation present in the NZW strain. We indeed showed that binding of macrophage complexes A, B and C to the full-length 3′-UTR cRNA probe TNF-9 containing a non-mutated ARE (Fig. 3, lane 2) was more efficient than the binding to the TNF-9m cRNA probe containing mutated ARE from NZW TNFα 3′-UTR. The binding of macrophage proteins to the 3′-UTR of TNFα mRNA from NZW mice was therefore hindered by a GAU insertional mutation in the first main ARE. The effects of nucleotide insertions in the main ARE of the 3′-UTR of TNFα mRNA was further documented by performing RNA-binding assays with synthetic cRNA encoding the first main 39 nucleotide protein-binding region with or without trinucleotide (GAU or CAU) insertions in the main ARE. We showed that the binding of macrophage complexes B and C to the TNF-10m cRNA probe, as well as to the TNF-10m2 cRNA probe was hindered by mutational insertions in the main ARE (Figs 4A and 5A; Table 1). These mutations therefore influence the binding pattern of macrophage complexes to sequences that are believed to post-transcriptionally regulate TNFα. The levels of complex B and C obtained with the TNF-10m probe (with GAU insertion) are very similar to what is seen when the TNF-10m2 probe (CAU insertion) was employed in the assay (Table 1). Furthermore, we demonstrated using SDS–PAGE gel analysis that both trinucleotide and dinucleotide insertions (data not shown) hinder the binding of several RNA-binding proteins of apparent molecular weights 48, 81, 89 and 125 kDa to the main ARE (Figs 4B and 5B). A GAU trinucleotide insertion in the main ARE (as observed in NZW mice known as low producers of TNFα) hinders the binding of macrophage proteins to a region of the mRNA that regulates the translational repression and inducibility of TNFα. Therefore, it is possible that the decreased levels of these proteins may be the cause of decreased TNFα production.
There is a large family of RNA-binding proteins involved in the post-transcriptional regulation of cytokines containing AREs in the 3′-UTR of their mRNA. Results published by Bohjanen et al. (19) indicate that two proteins present in T cells are capable of binding to the AREs present in the 3′-UTR of IL-2, TNFα and GM-CSF mRNA. They show that one of the proteins (AU-A) is present in the nucleus of unstimulated T cells, while the other is a cytoplasmic protein (AU-B) that is rapidly induced by stimulation of the T-cell receptor-CD3 complex. AU-A, unlike AU-B, can also bind to AREs present in the 3′-UTR of protooncogenes. The increase in binding of AU-B (due to CD3-T cell receptor mediated stimulation of T cells) results in decreased binding of AU-A. Furthermore, they show that binding of AU-B to the ARE is inversely correlated with stabilization of GM-CSF mRNA. Katz et al. (21) showed that AU-A is capable of shuttling between the nucleus and the cytoplasm. They indicate that AU-A may be a component of a complex involved in the transport of mRNA from the nucleus to the cytoplasm. These two complexes therefore seem to play important roles in the post-transcriptional regulation of cytokines containing ARE in their 3′-UTR. We have shown previously that three complexes A, B and C are capable of binding to the main ARE present in the 3′-UTR of TNFα mRNA (22,23). Complexes B and C (similarly to AU-A) are predominately found in the nucleus, while complex A is localized predominately to the cytoplasm (similarly to AU-B). Binding of complexes B and C, (unlike complex A) to the main ARE of the TNFα 3′-UTR is competed by ARE present in the 3′-UTR of c-fos. These two complexes, therefore, can bind to both cytokine and protooncogene mRNA containing ARE in their 3′-UTR. Although predominant in the nucleus, complex C is also present in the cytoplasm associated with polyribosomal supercomplexes in macrophages (data not shown). We therefore hypothesize that complex C, similarly to AU-A in T cells, is involved in the transport of RNA from nucleus to cytoplasm in macrophages. Several proteins capable of binding to the main ARE of the TNFα mRNA 3′-UTR have recently been identified in both macrophage and non-macrophage cells. Some of these proteins are the ELAV-like proteins (36). These proteins have been shown to be involved in the regulation of stability and translatability of ARE containing mRNAs (53–55). Antic and Keene (48) have reported that ELAV protein–RNA complexes are associated with polyribosomes, and postulated that this protein may be involved in mRNA transport. HuR, a member of the ELAV-like proteins, has been shown to play a crucial role in the post-transcriptional regulation of mRNA containing ARE such as c-fos (a protooncogene) and iNOS II (enzyme involved in the production of NO) (49,50,56). Peng et al. (49) have shown that HuR is able to bind to ARE in the 3′-UTR of c-fos and that overexpression of this protein in NIH 3T3 cells results in a decreased decay of transcripts containing c-fos ARE. They also demonstated that the formation of complexes containing HuR in the cytoplasm is increased when the cells are stimulated with Actinomycin D (49). HuR possesses a nuclear–cytoplasmic shuttling sequence that is believed to be important for the functioning of this protein as a stabilizer of message containing ARE (38). In our studies we have shown that complexes whose binding activity is hindered by a trinucleotide mutation in the 3′-UTR of TNFα mRNA are associated with polyribosomes in the cytoplasm (data not shown) and are influenced by inhibitors of transcriptional activity (Fig. 6A and B). These complexes are also able to bind to ARE in both protooncogenes and cytokines. We therefore hypothesized that these complexes may contain HuR. Supershift experiments using the TNF-10 cRNA probe and monoclonal antibodies directed against HuR allowed us to demonstrate that complexes B and C do indeed contain HuR (Fig. 7). The shifted complex was present to a much lesser extent when the assay was performed with the TNF-10m probe (in comparison to the TNF-10) (data not shown). Interestingly, HuR has a molecular weight of ∼37 kDa (56). Our SDS–PAGE studies indicate that a 48 kDa RNA–protein complex is dramatically affected by the mutation in the ARE (Figs 4B and 5B) as well as by Actinomycin D (Fig. 6B). Considering that the TNF-10 probe accounts for ∼10 kDa, the RNA–protein complex on the SDS–PAGE gel most likely contains HuR.
Overall, we demonstrated that the mutation affects the binding of a complex which contains a protein that is needed for the stability and transport of the TNFα message. We showed that an insertional mutation in the main ARE hinders binding of HuR (present in complex C) to the region in the TNFα message which mediates post-transcriptional regulation of TNFα. The decreased production of TNFα protein by peritoneal macrophages obtained from NZW mice may be due to the decreased transport of TNFα mRNA from the nucleus to the cytoplasm. The decreased binding of HuR to TNFα mRNA in the nucleus of macrophages in NZW mice may result in an increased turnover of message, and hence decreased transport of RNA to the polyribosomes (since complex C is associated with polyribosomes) where the mRNA is translated into protein, which would account for a decreased production of TNFα protein observed in NZW mice.
Acknowledgments
ACKNOWLEDGEMENTS
We thank A. Cerami for providing us with the murine TNFα cDNA, and S.Daly for reviewing of this manuscript. This work was supported by grants from the Medical Research Council of Canada and the Canadian Cystic Fibrosis Foundation to D.R. S.D. is a recipient of an MRC Doctoral Research Award. D.R. is a recipient of a FRSQ Chercheurs Nationaux Scholarship.
References
- 1.Vassalli P. (1992) The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol., 10, 411–452. [DOI] [PubMed] [Google Scholar]
- 2.Camussi G., Albano,E., Tetta,C. and Bussolino,F. (1991) The molecular action of tumor necrosis factor-alpha. Eur. J. Biochem., 202, 3–14. [DOI] [PubMed] [Google Scholar]
- 3.Ulich T.R., Shin,S.S. and del Castillo,J. (1993) Haematologic effects of TNF. Res. Immunol., 144, 347–354. [DOI] [PubMed] [Google Scholar]
- 4.Beutler B. and Cerami,A. (1988) Tumor necrosis, cachexia, shock, and inflammation: a common mediator. Annu. Rev. Biochem., 57, 505–518. [DOI] [PubMed] [Google Scholar]
- 5.Beutler B. (1995) TNF, immunity and inflammatory disease: lessons of the past decade. J. Invest. Med., 43, 227–235. [PubMed] [Google Scholar]
- 6.Strieter R.M., Kunkel,S.L. and Bone,R.C. (1993) Role of tumor necrosis factor-alpha in disease states and inflammation. Crit. Care Med., 21, S447–S463. [DOI] [PubMed] [Google Scholar]
- 7.Standiford T.J. and Strieter,R.M. (1992) TNF and IL-1 in sepsis: good cytokines gone bad. J. Lab. Clin. Med., 120, 179–180. [PubMed] [Google Scholar]
- 8.Biragyn A. and Nedospasov,S.A. (1995) Lipopolysaccharide-induced expression of TNF-alpha gene in the macrophage cell line ANA-1 is regulated at the level of transcription processivity. J. Immunol ., 155, 674–683. [PubMed] [Google Scholar]
- 9.Han J., Brown,T. and Beutler,B. (1990) Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level [published erratum appears in J. Exp. Med. (1990) 171, 971–972]. J. Exp. Med., 171, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beutler B. (1990) Regulation of cachectin biosynthesis occurs at multiple levels. Prog. Clin. Biol. Res., 349, 229–240. [PubMed] [Google Scholar]
- 11.Beutler B. and Brown,T. (1991) A CAT reporter construct allows ultrasensitive estimation of TNF synthesis, and suggests that the TNF gene has been silenced in non-macrophage cell lines. J. Clin. Invest., 87, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J. and Beutler,B. (1990) The essential role of the UA-rich sequence in endotoxin-induced cachectin/TNF synthesis. Eur. Cytokine Netw., 1, 71–75. [PubMed] [Google Scholar]
- 13.Han J.H., Beutler,B. and Huez,G. (1991) Complex regulation of tumor necrosis factor mRNA turnover in lipopolysaccharide-activated macrophages. Biochim. Biophys. Acta, 1090, 22–28. [DOI] [PubMed] [Google Scholar]
- 14.Han J., Huez,G. and Beutler,B. (1991) Interactive effects of the tumor necrosis factor promoter and 3′-untranslated regions. J. Immunol ., 146, 1843–1848. [PubMed] [Google Scholar]
- 15.Greenberg M.E. and Belasco,J.G. (1993) In Belasco,J.G. and Brawerman,G. (eds), Control of Messenger RNA Stability. Academic Press, San Diego, CA, pp. 199–218.
- 16.Schiavi S.C., Belasco,J.G. and Greenberg,M.E. (1992) Regulation of proto-oncogene mRNA stability. Biochim. Biophys. Acta, 1114, 95–106. [DOI] [PubMed] [Google Scholar]
- 17.Caput D., Beutler,B., Hartog,K., Thayer,R., Brown-Shimer,S. and Cerami,A. (1986) Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc. Natl Acad. Sci. USA, 83, 1670–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruys V., Marinx,O., Shaw,G., Deschamps,J. and Huez,G. (1989) Translational blockade imposed by cytokine-derived UA-rich sequences. Science, 245, 852–855. [DOI] [PubMed] [Google Scholar]
- 19.Bohjanen P.R., Petryniak,B., June,C.H., Thompson,C.B. and Lindsten,T. (1992) AU RNA-binding factors differ in their binding specificities and affinities. J. Biol. Chem., 267, 6302–6309. [PubMed] [Google Scholar]
- 20.Zhang W., Wagner,B.J., Ehrenman,K., Schaefer,A.W., DeMaria,C.T., Crater,D., DeHaven,K., Long,L. and Brewer,G. (1993) Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell Biol., 13, 7652–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz D.A., Theodorakis,N.G., Cleveland,D.W., Lindsten,T. and Thompson,C.B. (1994) AU-A, an RNA-binding activity distinct from hnRNP A1, is selective for AUUUA repeats and shuttles between the nucleus and the cytoplasm. Nucleic Acids Res., 22, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hel Z., Skamene,E. and Radzioch,D. (1996) Two distinct regions in the 3′ untranslated region of tumor necrosis factor alpha mRNA form complexes with macrophage proteins. Mol. Cell Biol., 16, 5579–5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hel Z., Di Marco,S. and Radzioch,D. (1998) Characterization of the RNA binding proteins forming complexes with a novel putative regulatory region in the 3′-UTR of TNF-alpha mRNA. Nucleic Acids Res., 26, 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob C.O. and Tashman,N.B. (1993) Disruption in the AU motif of the mouse TNF-alpha 3′ UTR correlates with reduced TNF production by macrophages in vitro. Nucleic Acids Res., 21, 2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob C.O. and McDevitt,H.O. (1988) Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature, 331, 356–358. [DOI] [PubMed] [Google Scholar]
- 26.Jacob C.O., Hwang,F., Lewis,G.D. and Stall,A.M. (1991) Tumor necrosis factor alpha in murine systemic lupus erythematosus disease models: implications for genetic predisposition and immune regulation. Cytokine, 3, 551–561. [DOI] [PubMed] [Google Scholar]
- 27.Kalechman Y., Gafter,U., Da,J.P., Albeck,M., Alarcon-Segovia,D. and Sredni,B. (1997) Delay in the onset of systemic lupus erythematosus following treatment with the immunomodulator AS101: association with IL-10 inhibition and increase in TNF-alpha levels. J. Immunol ., 159, 2658–2667. [PubMed] [Google Scholar]
- 28.Theofilopoulos A.N. and Dixon,F.J. (1985) Murine models of systemic lupus erythematosus. Adv. Immunol., 37, 269–390. [DOI] [PubMed] [Google Scholar]
- 29.Gordon C., Ranges,G.E., Greenspan,J.S. and Wofsy,D. (1989) Chronic therapy with recombinant tumor necrosis factor-alpha in autoimmune NZB/NZW F1 mice. Clin. Immunol. Immunopathol., 52, 421–434. [DOI] [PubMed] [Google Scholar]
- 30.Jacob C.O., Lee,S.K. and Strassmann,G. (1996) Mutational analysis of TNF-alpha gene reveals a regulatory role for the 3′-untranslated region in the genetic predisposition to lupus-like autoimmune disease. J. Immunol ., 156, 3043–3050. [PubMed] [Google Scholar]
- 31.Beutler B. and Brown,T. (1993) Polymorphism of the mouse TNF-alpha locus: sequence studies of the 3′-untranslated region and first intron [published erratum appears in Gene (1993) 136, 379]. Gene, 129, 279–283. [DOI] [PubMed] [Google Scholar]
- 32.Gueydan C., Droogmans,L., Chalon,P., Huez,G., Caput,D. and Kruys,V. (1999) Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor alpha mRNA. J. Biol. Chem., 274, 2322–2326. [DOI] [PubMed] [Google Scholar]
- 33.Dember L.M., Kim,N.D., Liu,K.Q. and Anderson,P. (1996) Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem., 271, 2783–2788. [DOI] [PubMed] [Google Scholar]
- 34.Lai W.S., Carballo,E., Strum,J.R., Kennington,E.A., Phillips,R.S. and Blackshear,P.J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell Biol., 19, 4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carballo E., Lai,W.S. and Blackshear,P.J. (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science, 281, 1001–1005. [DOI] [PubMed] [Google Scholar]
- 36.Sakai K., Kitagawa,Y. and Hirose,G. (1999) Binding of neuronal ELAV-like proteins to the uridine-rich sequence in the 3′-untranslated region of tumor necrosis factor-alpha messenger RNA. FEBS Lett., 446, 157–162. [DOI] [PubMed] [Google Scholar]
- 37.Ma W.J., Cheng,S., Campbell,C., Wright,A. and Furneaux,H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem., 271, 8144–8151. [DOI] [PubMed] [Google Scholar]
- 38.Fan X.C. and Steitz,J.A. (1998) HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl Acad. Sci. USA, 95, 15293–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radzioch D., Hudson,T., Boule,M., Barrera,L., Urbance,J.W., Varesio,L. and Skamene,E. (1991) Genetic resistance/susceptibility to mycobacteria: phenotypic expression in bone marrow derived macrophage lines. J. Leukoc. Biol., 50, 263–272. [DOI] [PubMed] [Google Scholar]
- 40.Sheehan K.C., Ruddle,N.H. and Schreiber,R.D. (1989) Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J. Immunol., 142, 3884–3893. [PubMed] [Google Scholar]
- 41.Gurevich V.V., Pokrovskaya,I.D., Obukhova,T.A. and Zozulya,S.A. (1991) Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Anal. Biochem., 195, 207–213. [DOI] [PubMed] [Google Scholar]
- 42.Dignam J.D. (1990) Preparation of extracts from higher eukaryotes. Methods Enzymol., 182, 194–203. [DOI] [PubMed] [Google Scholar]
- 43.Fields A.P., Pettit,G.R. and May,W.S. (1988) Phosphorylation of lamin B at the nuclear membrane by activated protein kinase C. J. Biol. Chem., 263, 8253–8260. [PubMed] [Google Scholar]
- 44.Gueydan C., Houzet,L., Marchant,A., Sels,A., Huez,G. and Kruys,V. (1996) Engagement of tumor necrosis factor mRNA by an endotoxin-inducible cytoplasmic protein [published erratum appears in Mol. Med. (1996) 2, 786]. Mol. Med., 2, 479–488. [PMC free article] [PubMed] [Google Scholar]
- 45.Ozols J. (1990) Preparation of membrane fractions. Methods Enzymol., 182, 225–235. [DOI] [PubMed] [Google Scholar]
- 46.Wilson T. and Treisman,R. (1988) Removal of poly(A) and consequent degradation of c-fos mRNA facilitated by 3′ AU-rich sequences. Nature, 336, 396–399. [DOI] [PubMed] [Google Scholar]
- 47.Shyu A.B., Greenberg,M.E. and Belasco,J.G. (1989) The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev., 3, 60–72. [DOI] [PubMed] [Google Scholar]
- 48.Antic D. and Keene,J.D. (1998) Messenger ribonucleoprotein complexes containing human ELAV proteins: interactions with cytoskeleton and translational apparatus. J. Cell Sci., 111, 183–197. [DOI] [PubMed] [Google Scholar]
- 49.Peng S.S., Chen,C.Y., Xu,N. and Shyu,A.B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J., 17, 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X.C. and Steitz,J.A. (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J., 17, 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schook L.B., Albrecht,H., Gallay,P. and Jongeneel,C.V. (1994) Cytokine regulation of TNF-alpha mRNA and protein production by unprimed macrophages from C57Bl/6 and NZW mice. J. Leukoc. Biol., 56, 514–520. [DOI] [PubMed] [Google Scholar]
- 52.Bazzoni F. and Beutler,B. (1995) Comparative expression of TNF-alpha alleles from normal and autoimmune-prone MHC haplotypes. J. Inflamm., 45, 106–114. [PubMed] [Google Scholar]
- 53.Antic D. and Keene,J.D. (1997) Embryonic lethal abnormal visual RNA-binding proteins involved in growth, differentiation, and posttranscriptional gene expression. Am. J. Hum. Genet., 61, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain R.G., Andrews,L.G., McGowan,K.M., Pekala,P.H. and Keene,J.D. (1997) Ectopic expression of Hel-N1, an RNA-binding protein, increases glucose transporter (GLUT1) expression in 3T3-L1 adipocytes. Mol. Cell Biol., 17, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Myer V.E., Fan,X.C. and Steitz,J.A. (1997) Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J., 16, 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez-Pascual F., Hausding,M., Ihrig-Biedert,I., Furneaux,H., Levy,A.P., Forstermann,U. and Kleinert,H. (2000) Complex contribution of the 3′-untranslated region to the expressional regulation of the human inducible nitric-oxide synthase gene. Involvement of the RNA-binding protein HuR. J. Biol. Chem., 275, 26040–26049. [DOI] [PubMed] [Google Scholar]