

Abstract
In the title compound, C10H13NO, the dihedral angle between the amide group and the phenyl ring is 30.0 (3)°. In the crystal structure, intermolecular N—H⋯O hydrogen bonds link molecules into one-dimensional chains along the a axis.
Related literature
For related literature, see: Clayden et al. (2006 ▶); Kopka et al. (2005 ▶); Smart (2001 ▶); Van Waarde et al. (2004 ▶); Stephenson, Wilson et al. (2008 ▶); Stephenson, van Oosten et al. (2008 ▶).
Experimental
Crystal data
C10H13NO
M r = 163.21
Monoclinic,
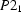
a = 5.0093 (7) Å
b = 10.1250 (13) Å
c = 9.6714 (14) Å
β = 104.133 (7)°
V = 475.68 (11) Å3
Z = 2
Mo Kα radiation
μ = 0.07 mm−1
T = 150 (1) K
0.14 × 0.13 × 0.08 mm
Data collection
Bruker Nonius KappaCCD diffractometer
Absorption correction: multi-scan (SORTAV; Blessing 1995 ▶) T min = 0.954, T max = 0.996
2462 measured reflections
887 independent reflections
621 reflections with I > 2σ(I)
R int = 0.061
Refinement
R[F 2 > 2σ(F 2)] = 0.057
wR(F 2) = 0.148
S = 1.06
887 reflections
114 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.18 e Å−3
Δρmin = −0.21 e Å−3
Data collection: COLLECT (Nonius, 2002 ▶); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997 ▶); data reduction: DENZO-SMN; program(s) used to solve structure: SIR92 (Altomare et al., 1994 ▶); program(s) used to refine structure: SHELXTL (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2003 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808012804/hb2729sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808012804/hb2729Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N⋯O1i | 0.83 (5) | 2.10 (5) | 2.890 (5) | 160 (5) |
Symmetry code: (i) 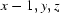 .
.
Acknowledgments
The authors thank Dr Karin A. Stephenson, Dr Andrei K. Yudin and Dr Alan A. Wilson for helpful discussions. We thank Dr Sylvain Houle for allowing the CAMH PET Centre facilities to be used for this research. Financial support for this work was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes for Health Research in the form of a Collaborative Health Research Projects Grant (CHRPJ 322787-06).
supplementary crystallographic information
Comment
The isopropylamine moiety is a common structural feature in many pharmaceutical compounds, in particular among β-adrenergic receptor antagonists (β-blockers) (Van Waarde et al., 2004; Kopka et al., 2005). Recent work in our laboratory (Stephenson, Wilson et al., 2008; Stephenson, van Oosten et al., 2008) and others (Van Waarde et al., 2004; Kopka et al., 2005) has focused on developing β-blockers labeled with the positron emitting isotope fluorine-18 (t1/2 = 109.7 min) at the isopropyl moiety for medical imaging with positron emission tomography. It is established that substitution of fluorine into a drug often enhances its biological properties (Smart, 2001). Our goal is to structurally characterize the isopropylamine group for comparison with fluorinated analogs developed in our laboratory. Herein we report the single-crystal X-ray structure of the title compound, (I), (Fig. 1).
The dihedral angle between the essentially planar set of atoms C7/O1/N1/C8 [r.m.s. deviation 0.006 Å] and the benzene ring (C1–C6) in (I) is 30.0 (3)°. In the crystal structure, intermolecular N—H···O hydrogen bonds link molecules into one-dimensional chains along the a axis (Table 1, Fig. 2).
Experimental
N-Isopropylbenzamide was made according to a literature procedure (Clayden et al., 2006), with minor modifications. Benzoyl chloride (0.825 ml, 7.11 mmol) was added to CH2Cl2 (17 ml, 0.4 M) under nitrogen. The mixture was cooled in an ice bath to 273 K and stirred for 10 min. Isopropylamine (1.8 ml, 21.33 mmol) was added dropwise. Upon completion of this addition the ice bath was removed and the reaction mixture was stirred at room temperature for 1.5 h. When the starting material was consumed (monitored by TLC) the reaction mixture was diluted with H2O (150 ml), extracted with CH2Cl2 (3 × 50 ml), washed with H2O (2 × 100 ml) followed by brine (2 × 100 ml), dried over Na2SO4, and concentrated. No further purification was necessary. Colourless blocks of (I) were obtained by slow evaporation of a solution of the title compound in CDCl3. 1H NMR (CDCl3, 300 MHz) d = 7.78–7.67 (m, 2H), 7.51–7.36 (m, 3H), 5.99 (br, 1H), 4.37–4.18 (m, 1H), 1.25 (d, J = 6.5 Hz, 6H) 13C NMR (CDCl3, 75 MHz) d = 166.9, 135.2, 131.5, 128.7, 127.0, 42.1, 23.1.
Refinement
In the absence of significant anamlous dispersion effects, Friedel pairs were merged before refinement. The H atoms bonded to C atoms were placed in calculated positions with C—H = 0.95 Å and 0.98 Å (methyl). They were included in the refinement in the riding-model approximation with Uiso(H) = 1.2Ueq(C) or Uiso(H) = 1.5Ueq(C) for methyl H atoms. The position of the H atom bonded to the N atom was refined independently with Uiso(H) = 1.2Ueq(N).
Figures
Fig. 1.

The molecular structure of (I) showing 30% probability displacement ellipsoids (arbitrary spheres for H atoms).
Fig. 2.

Part of the crystal structure of (I) showing hydrogen bonds as dashed lines.
Crystal data
| C10H13NO | F000 = 176 |
| Mr = 163.21 | Dx = 1.140 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: P 2yb | Cell parameters from 2462 reflections |
| a = 5.0093 (7) Å | θ = 3.0–25.0º |
| b = 10.1250 (13) Å | µ = 0.07 mm−1 |
| c = 9.6714 (14) Å | T = 150 (1) K |
| β = 104.133 (7)º | Block, colourless |
| V = 475.68 (11) Å3 | 0.14 × 0.13 × 0.08 mm |
| Z = 2 |
Data collection
| Bruker Nonius KappaCCD diffractometer | 887 independent reflections |
| Radiation source: fine-focus sealed tube | 621 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.061 |
| Detector resolution: 9 pixels mm-1 | θmax = 25.0º |
| T = 150(1) K | θmin = 3.0º |
| φ scans and ω scans with κ offsets | h = −5→5 |
| Absorption correction: multi-scan(SORTAV; Blessing 1995) | k = −12→10 |
| Tmin = 0.954, Tmax = 0.996 | l = −11→11 |
| 2462 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.057 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.148 | w = 1/[σ2(Fo2) + (0.0784P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max < 0.001 |
| 887 reflections | Δρmax = 0.18 e Å−3 |
| 114 parameters | Δρmin = −0.21 e Å−3 |
| 1 restraint | Extinction correction: none |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.6751 (6) | 0.1703 (3) | 0.8045 (3) | 0.0424 (8) | |
| N1 | 0.2696 (8) | 0.1717 (4) | 0.8673 (4) | 0.0411 (10) | |
| H1N | 0.103 (11) | 0.188 (5) | 0.839 (5) | 0.049* | |
| C1 | 0.3226 (9) | 0.3127 (4) | 0.6749 (5) | 0.0350 (11) | |
| C2 | 0.4247 (10) | 0.3165 (5) | 0.5544 (5) | 0.0452 (13) | |
| H2A | 0.5601 | 0.2545 | 0.5432 | 0.054* | |
| C3 | 0.3286 (11) | 0.4116 (6) | 0.4493 (5) | 0.0535 (14) | |
| H3A | 0.3957 | 0.4126 | 0.3655 | 0.064* | |
| C4 | 0.1377 (10) | 0.5034 (5) | 0.4666 (6) | 0.0516 (14) | |
| H4A | 0.0734 | 0.5682 | 0.3951 | 0.062* | |
| C5 | 0.0391 (11) | 0.5014 (5) | 0.5883 (6) | 0.0523 (14) | |
| H5A | −0.0912 | 0.5656 | 0.6010 | 0.063* | |
| C6 | 0.1296 (9) | 0.4064 (5) | 0.6915 (5) | 0.0451 (13) | |
| H6A | 0.0597 | 0.4051 | 0.7744 | 0.054* | |
| C7 | 0.4356 (8) | 0.2117 (4) | 0.7864 (4) | 0.0362 (12) | |
| C8 | 0.3461 (10) | 0.0743 (5) | 0.9808 (5) | 0.0474 (13) | |
| H8A | 0.5489 | 0.0586 | 0.9996 | 0.057* | |
| C9 | 0.1984 (13) | −0.0561 (5) | 0.9351 (6) | 0.0648 (16) | |
| H9A | 0.2525 | −0.0903 | 0.8512 | 0.097* | |
| H9B | −0.0011 | −0.0417 | 0.9116 | 0.097* | |
| H9C | 0.2488 | −0.1200 | 1.0132 | 0.097* | |
| C10 | 0.2833 (11) | 0.1271 (6) | 1.1160 (5) | 0.0556 (15) | |
| H10A | 0.3743 | 0.2126 | 1.1400 | 0.083* | |
| H10B | 0.3510 | 0.0647 | 1.1942 | 0.083* | |
| H10C | 0.0839 | 0.1380 | 1.1013 | 0.083* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0319 (17) | 0.0442 (18) | 0.0515 (17) | 0.0024 (15) | 0.0112 (13) | 0.0032 (16) |
| N1 | 0.0326 (19) | 0.046 (2) | 0.047 (2) | 0.003 (2) | 0.0149 (18) | 0.012 (2) |
| C1 | 0.033 (2) | 0.035 (2) | 0.039 (3) | −0.006 (2) | 0.012 (2) | 0.000 (2) |
| C2 | 0.049 (3) | 0.044 (3) | 0.046 (3) | 0.001 (2) | 0.019 (2) | 0.003 (2) |
| C3 | 0.057 (3) | 0.061 (3) | 0.046 (3) | −0.003 (3) | 0.019 (3) | 0.012 (3) |
| C4 | 0.049 (3) | 0.051 (3) | 0.050 (3) | −0.002 (3) | 0.004 (3) | 0.020 (3) |
| C5 | 0.053 (3) | 0.040 (3) | 0.066 (3) | 0.007 (3) | 0.017 (3) | 0.013 (3) |
| C6 | 0.046 (3) | 0.042 (3) | 0.051 (3) | 0.003 (2) | 0.017 (2) | 0.005 (3) |
| C7 | 0.035 (3) | 0.034 (3) | 0.037 (2) | −0.004 (2) | 0.005 (2) | −0.008 (2) |
| C8 | 0.041 (3) | 0.054 (3) | 0.048 (3) | 0.012 (3) | 0.012 (2) | 0.017 (3) |
| C9 | 0.085 (4) | 0.043 (3) | 0.064 (3) | 0.010 (3) | 0.014 (3) | 0.012 (3) |
| C10 | 0.062 (3) | 0.062 (4) | 0.044 (3) | −0.001 (3) | 0.015 (2) | 0.012 (3) |
Geometric parameters (Å, °)
| O1—C7 | 1.242 (5) | C5—C6 | 1.380 (7) |
| N1—C7 | 1.337 (6) | C5—H5A | 0.9500 |
| N1—C8 | 1.455 (6) | C6—H6A | 0.9500 |
| N1—H1N | 0.83 (5) | C8—C10 | 1.516 (7) |
| C1—C2 | 1.383 (6) | C8—C9 | 1.525 (8) |
| C1—C6 | 1.392 (6) | C8—H8A | 1.0000 |
| C1—C7 | 1.493 (6) | C9—H9A | 0.9800 |
| C2—C3 | 1.398 (8) | C9—H9B | 0.9800 |
| C2—H2A | 0.9500 | C9—H9C | 0.9800 |
| C3—C4 | 1.373 (7) | C10—H10A | 0.9800 |
| C3—H3A | 0.9500 | C10—H10B | 0.9800 |
| C4—C5 | 1.384 (7) | C10—H10C | 0.9800 |
| C4—H4A | 0.9500 | ||
| C7—N1—C8 | 124.0 (4) | O1—C7—N1 | 122.2 (4) |
| C7—N1—H1N | 118 (4) | O1—C7—C1 | 121.0 (4) |
| C8—N1—H1N | 117 (4) | N1—C7—C1 | 116.7 (4) |
| C2—C1—C6 | 119.2 (4) | N1—C8—C10 | 109.9 (4) |
| C2—C1—C7 | 118.3 (4) | N1—C8—C9 | 110.4 (4) |
| C6—C1—C7 | 122.4 (4) | C10—C8—C9 | 111.5 (4) |
| C1—C2—C3 | 120.0 (5) | N1—C8—H8A | 108.3 |
| C1—C2—H2A | 120.0 | C10—C8—H8A | 108.3 |
| C3—C2—H2A | 120.0 | C9—C8—H8A | 108.3 |
| C4—C3—C2 | 120.3 (5) | C8—C9—H9A | 109.5 |
| C4—C3—H3A | 119.9 | C8—C9—H9B | 109.5 |
| C2—C3—H3A | 119.9 | H9A—C9—H9B | 109.5 |
| C3—C4—C5 | 119.8 (5) | C8—C9—H9C | 109.5 |
| C3—C4—H4A | 120.1 | H9A—C9—H9C | 109.5 |
| C5—C4—H4A | 120.1 | H9B—C9—H9C | 109.5 |
| C6—C5—C4 | 120.2 (5) | C8—C10—H10A | 109.5 |
| C6—C5—H5A | 119.9 | C8—C10—H10B | 109.5 |
| C4—C5—H5A | 119.9 | H10A—C10—H10B | 109.5 |
| C5—C6—C1 | 120.5 (4) | C8—C10—H10C | 109.5 |
| C5—C6—H6A | 119.8 | H10A—C10—H10C | 109.5 |
| C1—C6—H6A | 119.8 | H10B—C10—H10C | 109.5 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1i | 0.83 (5) | 2.10 (5) | 2.890 (5) | 160 (5) |
Symmetry codes: (i) x−1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB2729).
References
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst.27, 435.
- Blessing, R. H. (1995). Acta Cryst. A51, 33–38. [DOI] [PubMed]
- Clayden, J., Stimson, C. C. & Keenan, M. (2006). Chem. Commun.13, 1393–1394. [DOI] [PubMed]
- Kopka, K., Kaw, M. P., Breyholz, H. J., Faust, A., Hoeltke, C., Riemann, B., Schober, O., Schaefers, M. & Wagner, S. (2005). Curr. Med. Chem.12, 2057–2074. [DOI] [PubMed]
- Nonius (2002). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Smart, B. E. (2001). J. Fluorine Chem.109, 3–11.
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Stephenson, K. A., van Oosten, E. M., Wilson, A. A., Meyer, J. H., Houle, S. & Vasdev, N. (2008). Neurochem. Int. Accepted. [DOI] [PubMed]
- Stephenson, K. A., Wilson, A. A., Meyer, J. H., Houle, S. & Vasdev, N. (2008). J. Med. Chem. In the press. [DOI] [PubMed]
- Van Waarde, A., Vaalburg, W., Doze, P., Bosker, F. J. & Elsinga, P. H. (2004). Curr. Pharm. Des.10, 1519–1536. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808012804/hb2729sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808012804/hb2729Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report