

Abstract
Angiokeratoma corporis diffusum with glycopeptiduria is a recently recognized inborn error of glycoprotein catabolism resulting from the deficient activity of human alpha-N-acetylgalactosaminidase (E.C. 3.2.1.49; alpha-GalNAc). The first patient with this autosomal recessive disorder, a 46-yr-old consanguineous Japanese woman, presented with diffuse angiokeratoma, mild intellectual impairment, and peripheral neuroaxonal degeneration. Deficient alpha-GalNAc activity also has been reported in consanguineous brothers with an infantile-onset form of neuroaxonal dystrophy resulting from a missense mutation (designated E325K) in the alpha-GalNAc gene. To identify the mutation causing the phenotypically distinct adult-onset disorder, Southern and Northern hybridization analyses of DNA and RNA from the affected homozygote were performed which revealed a grossly normal alpha-GalNAc gene structure and normal transcript size and abundancy. Reverse transcription, amplification, and sequencing of the alpha-GalNAc transcript identified a single C to T transition at nucleotide (nt) 985 that predicted an arginine to tryptophan substitution in residue 329 (designated R329W) of the alpha-GalNAc polypeptide. This base substitution was confirmed by hybridization of PCR-amplified genomic DNA from family members with allele-specific oligonucleotides. Transient expression of an alpha-GalNAc construct containing the R329W mutation resulted in the expression of an immunoreactive polypeptide which had no detectable alpha-GalNAc activity. Comparison of the biosynthesis and stabilities of the transiently expressed and radiolabeled normal, E325K (infantile-onset) and R329W (adult-onset) alpha-GalNAc polypeptides in COS-1 cells indicated that both the mutant precursors were processed to the mature form; however, the E325K mutant polypeptide was more rapidly degraded than the R329W subunit, thereby providing a basis for the distinctly different infantile- and adult-onset phenotypes.
Full text
PDF



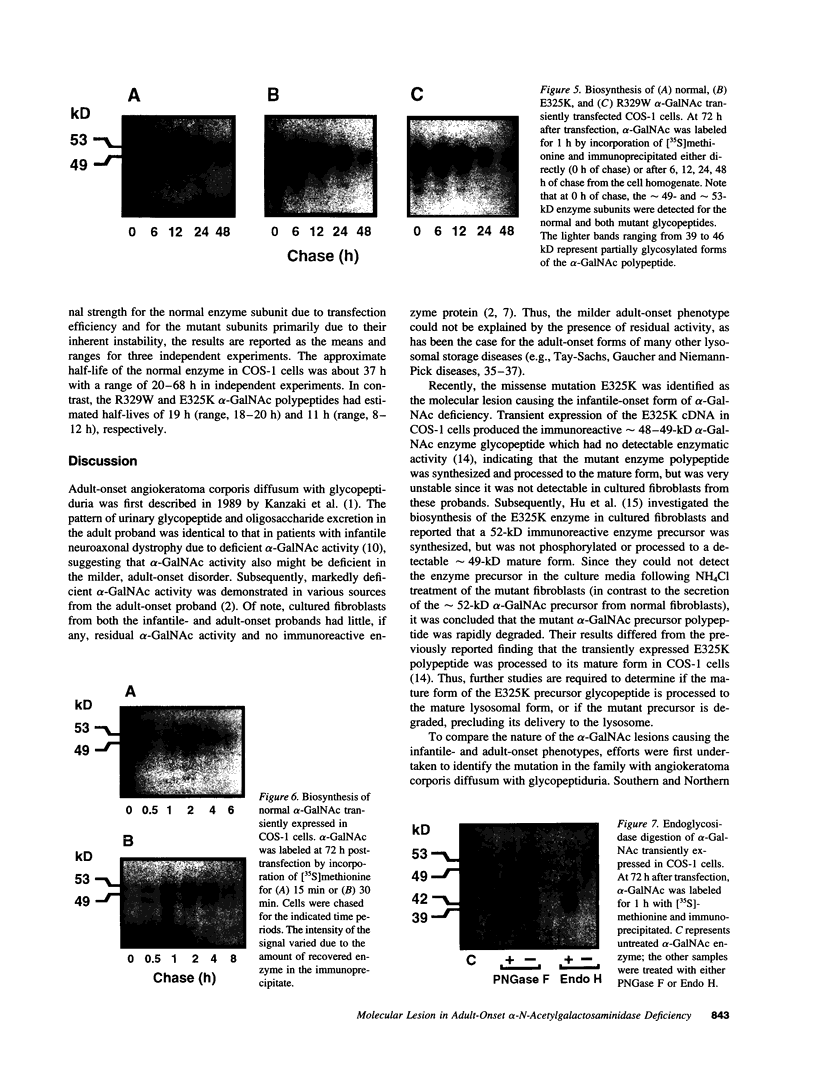


Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aldridge J., Kunkel L., Bruns G., Tantravahi U., Lalande M., Brewster T., Moreau E., Wilson M., Bromley W., Roderick T. A strategy to reveal high-frequency RFLPs along the human X chromosome. Am J Hum Genet. 1984 May;36(3):546–564. [PMC free article] [PubMed] [Google Scholar]
- Bernstein H. S., Bishop D. F., Astrin K. H., Kornreich R., Eng C. M., Sakuraba H., Desnick R. J. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J Clin Invest. 1989 Apr;83(4):1390–1399. doi: 10.1172/JCI114027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D. F., Desnick R. J. Affinity purification of alpha-galactosidase A from human spleen, placenta, and plasma with elimination of pyrogen contamination. Properties of the purified splenic enzyme compared to other forms. J Biol Chem. 1981 Feb 10;256(3):1307–1316. [PubMed] [Google Scholar]
- Chen C., Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987 Aug;7(8):2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Chou P. Y., Fasman G. D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- Desnick R. J. Gaucher disease: a century of delineation and understanding. Prog Clin Biol Res. 1982;95:1–30. [PubMed] [Google Scholar]
- Garnier J., Osguthorpe D. J., Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. 1978 Mar 25;120(1):97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y., Matsumoto Y., Matsumoto M., Toida T., Iida N., Matsubara T., Kanzaki T., Yokota M., Ishizuka I. Isolation and characterization of major urinary amino acid O-glycosides and a dipeptide O-glycoside from a new lysosomal storage disorder (Kanzaki disease). Excessive excretion of serine- and threonine-linked glycan in the patient urine. J Biol Chem. 1990 Jan 25;265(3):1693–1701. [PubMed] [Google Scholar]
- Hu P., Reuser A. J., Janse H. C., Kleijer W. J., Schindler D., Sakuraba H., Tsuji A., Suzuki Y., van Diggelen O. P. Biosynthesis of human alpha-N-acetylgalactosaminidase: defective phosphorylation and maturation in infantile alpha-NAGA deficiency. Biochem Biophys Res Commun. 1991 Mar 29;175(3):1097–1103. doi: 10.1016/0006-291x(91)91678-6. [DOI] [PubMed] [Google Scholar]
- Kanzaki T., Wang A. M., Desnick R. J. Lysosomal alpha-N-acetylgalactosaminidase deficiency, the enzymatic defect in angiokeratoma corporis diffusum with glycopeptiduria. J Clin Invest. 1991 Aug;88(2):707–711. doi: 10.1172/JCI115357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki T., Yokota M., Irie F., Hirabayashi Y., Wang A. M., Desnick R. J. Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal alpha-N-acetylgalactosaminidase activity. Clinical, morphologic, and biochemical studies. Arch Dermatol. 1993 Apr;129(4):460–465. [PubMed] [Google Scholar]
- Kanzaki T., Yokota M., Mizuno N., Matsumoto Y., Hirabayashi Y. Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum. Lancet. 1989 Apr 22;1(8643):875–877. doi: 10.1016/s0140-6736(89)92867-5. [DOI] [PubMed] [Google Scholar]
- Lehrach H., Diamond D., Wozney J. M., Boedtker H. RNA molecular weight determinations by gel electrophoresis under denaturing conditions, a critical reexamination. Biochemistry. 1977 Oct 18;16(21):4743–4751. doi: 10.1021/bi00640a033. [DOI] [PubMed] [Google Scholar]
- Linden H. U., Klein R. A., Egge H., Peter-Katalinic J., Dabrowski J., Schindler D. Isolation and structural characterization of sialic-acid-containing glycopeptides of the O-glycosidic type from the urine of two patients with an hereditary deficiency in alpha-N-acetylgalactosaminidase activity. Biol Chem Hoppe Seyler. 1989 Jul;370(7):661–672. doi: 10.1515/bchm3.1989.370.2.661. [DOI] [PubMed] [Google Scholar]
- Messing J., Vieira J. A new pair of M13 vectors for selecting either DNA strand of double-digest restriction fragments. Gene. 1982 Oct;19(3):269–276. doi: 10.1016/0378-1119(82)90016-6. [DOI] [PubMed] [Google Scholar]
- Myerowitz R., Costigan F. C. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J Biol Chem. 1988 Dec 15;263(35):18587–18589. [PubMed] [Google Scholar]
- Myerowitz R. Splice junction mutation in some Ashkenazi Jews with Tay-Sachs disease: evidence against a single defect within this ethnic group. Proc Natl Acad Sci U S A. 1988 Jun;85(11):3955–3959. doi: 10.1073/pnas.85.11.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navon R., Proia R. L. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989 Mar 17;243(4897):1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- Saiki R. K., Scharf S., Faloona F., Mullis K. B., Horn G. T., Erlich H. A., Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985 Dec 20;230(4732):1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- Sanger F., Coulson A. R., Barrell B. G., Smith A. J., Roe B. A. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J Mol Biol. 1980 Oct 25;143(2):161–178. doi: 10.1016/0022-2836(80)90196-5. [DOI] [PubMed] [Google Scholar]
- Schindler D., Bishop D. F., Wolfe D. E., Wang A. M., Egge H., Lemieux R. U., Desnick R. J. Neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. N Engl J Med. 1989 Jun 29;320(26):1735–1740. doi: 10.1056/NEJM198906293202606. [DOI] [PubMed] [Google Scholar]
- Schindler D., Kanzaki T., Desnick R. J. A method for the rapid detection of urinary glycopeptides in alpha-N-acetylgalactosaminidase deficiency and other lysosomal storage diseases. Clin Chim Acta. 1990 Sep;190(1-2):81–91. doi: 10.1016/0009-8981(90)90282-w. [DOI] [PubMed] [Google Scholar]
- Schägger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987 Nov 1;166(2):368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Southern E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975 Nov 5;98(3):503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Sweeley C. C., Ledonne N. C., Jr, Robbins P. W. Post-translational processing reactions involved in the biosynthesis of lysosomal alpha-N-acetylgalactosaminidase in cultured human fibroblasts. Arch Biochem Biophys. 1983 May;223(1):158–165. doi: 10.1016/0003-9861(83)90581-7. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Suchi M., Desnick R. J., Takada G., Schuchman E. H. Identification and expression of five mutations in the human acid sphingomyelinase gene causing types A and B Niemann-Pick disease. Molecular evidence for genetic heterogeneity in the neuronopathic and non-neuronopathic forms. J Biol Chem. 1992 Jun 25;267(18):12552–12558. [PubMed] [Google Scholar]
- Tarentino A. L., Trimble R. B., Plummer T. H., Jr Enzymatic approaches for studying the structure, synthesis, and processing of glycoproteins. Methods Cell Biol. 1989;32:111–139. doi: 10.1016/s0091-679x(08)61169-3. [DOI] [PubMed] [Google Scholar]
- Theophilus B., Latham T., Grabowski G. A., Smith F. I. Gaucher disease: molecular heterogeneity and phenotype-genotype correlations. Am J Hum Genet. 1989 Aug;45(2):212–225. [PMC free article] [PubMed] [Google Scholar]
- Wang A. M., Bishop D. F., Desnick R. J. Human alpha-N-acetylgalactosaminidase-molecular cloning, nucleotide sequence, and expression of a full-length cDNA. Homology with human alpha-galactosidase A suggests evolution from a common ancestral gene. J Biol Chem. 1990 Dec 15;265(35):21859–21866. [PubMed] [Google Scholar]
- Wang A. M., Desnick R. J. Structural organization and complete sequence of the human alpha-N-acetylgalactosaminidase gene: homology with the alpha-galactosidase A gene provides evidence for evolution from a common ancestral gene. Genomics. 1991 May;10(1):133–142. doi: 10.1016/0888-7543(91)90493-x. [DOI] [PubMed] [Google Scholar]
- Wang A. M., Schindler D., Desnick R. Schindler disease: the molecular lesion in the alpha-N-acetylgalactosaminidase gene that causes an infantile neuroaxonal dystrophy. J Clin Invest. 1990 Nov;86(5):1752–1756. doi: 10.1172/JCI114901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf H., Modrow S., Motz M., Jameson B. A., Hermann G., Förtsch B. An integrated family of amino acid sequence analysis programs. Comput Appl Biosci. 1988 Mar;4(1):187–191. doi: 10.1093/bioinformatics/4.1.187. [DOI] [PubMed] [Google Scholar]
- Wong G. G., Witek J. S., Temple P. A., Wilkens K. M., Leary A. C., Luxenberg D. P., Jones S. S., Brown E. L., Kay R. M., Orr E. C. Human GM-CSF: molecular cloning of the complementary DNA and purification of the natural and recombinant proteins. Science. 1985 May 17;228(4701):810–815. doi: 10.1126/science.3923623. [DOI] [PubMed] [Google Scholar]
- van Diggelen O. P., Schindler D., Kleijer W. J., Huijmans J. M., Galjaard H., Linden H. U., Peter-Katalinic J., Egge H., Dabrowski U., Cantz M. Lysosomal alpha-N-acetylgalactosaminidase deficiency: a new inherited metabolic disease. Lancet. 1987 Oct 3;2(8562):804–804. doi: 10.1016/s0140-6736(87)92542-6. [DOI] [PubMed] [Google Scholar]
- van Diggelen O. P., Schindler D., Willemsen R., Boer M., Kleijer W. J., Huijmans J. G., Blom W., Galjaard H. alpha-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. J Inherit Metab Dis. 1988;11(4):349–357. doi: 10.1007/BF01800424. [DOI] [PubMed] [Google Scholar]